
Pharmacokinetic Evaluation of New Drugs Using a Multi-Labelling Approach and PET Imaging: Application to a Drug Candidate with Potential Application in Neuromuscular Disorders
 , , , , ,
, , , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry




2.2. Biological Evaluation
2.2.1. Competitive Ligand Binding Assay of AHK2
2.2.2. In Situ Proximity Ligation Assay (PLA)
2.3. Radiochemistry
2.3.1. Radiolabelling with carbon-11
2.3.2. Radiolabelling with hydrogen-3
2.4. In Vitro Studies
In Vitro Metabolism
2.5. In Vivo Studies
2.5.1. General Considerations
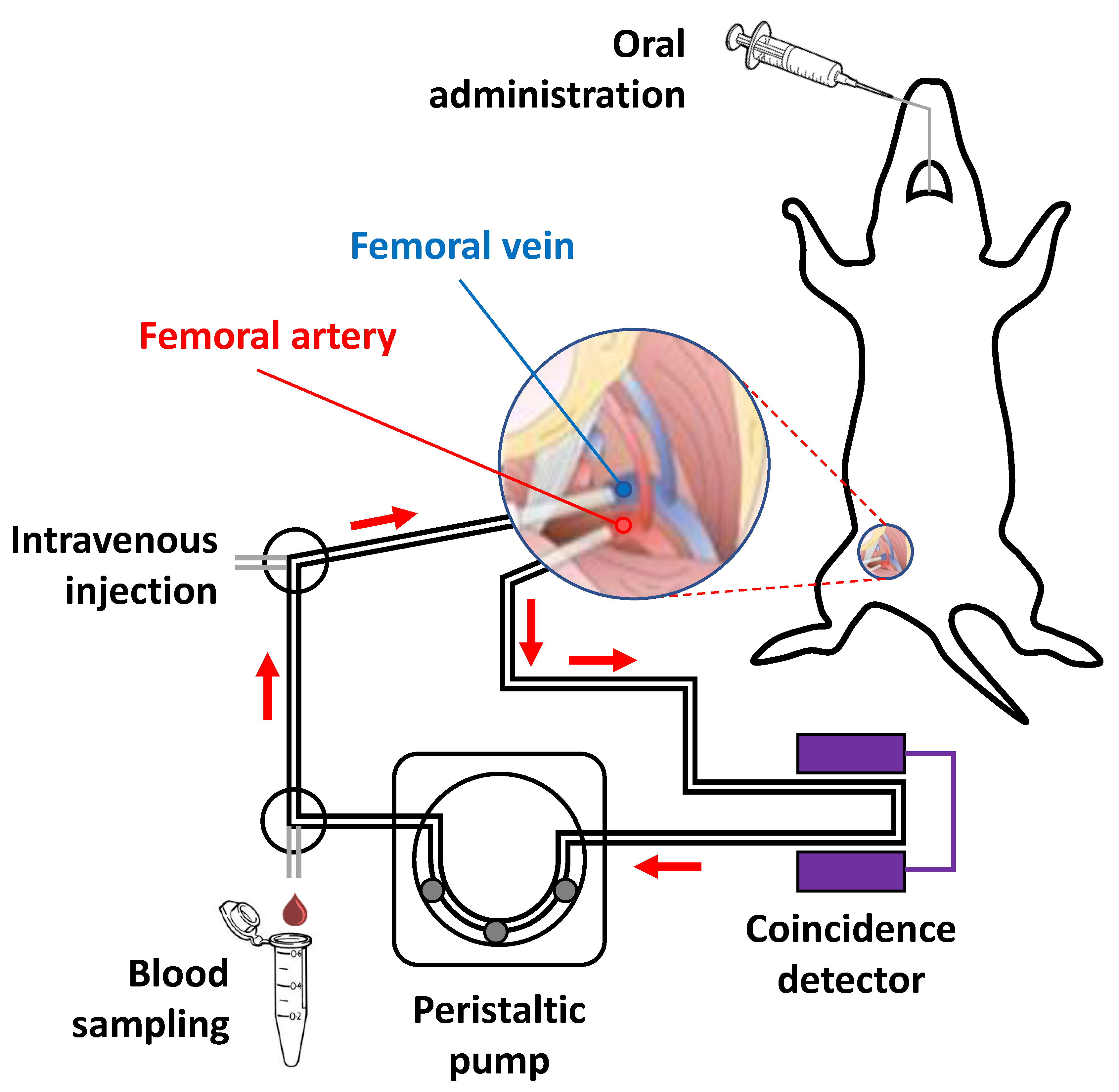
2.5.2. Extracorporeal Blood Circulation: Animal Surgery
2.5.3. Administration of Labelled Compounds, Blood Sampling and Determination of In Vivo Metabolism
2.5.4. Pharmacokinetic Analysis
2.5.5. PET Biodistribution Studies
3. Results
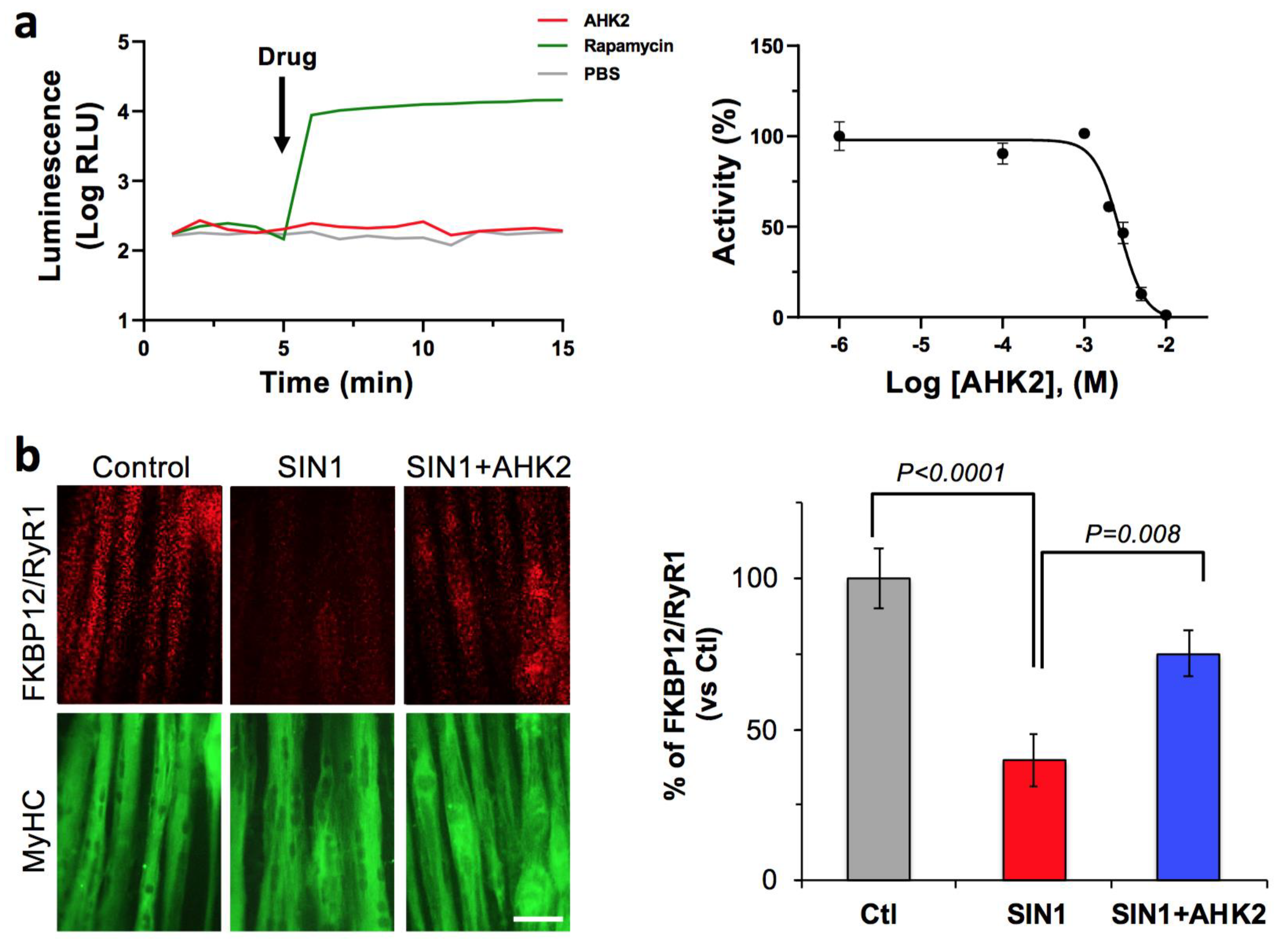
3.1. AHK2 Mechanisms of Action
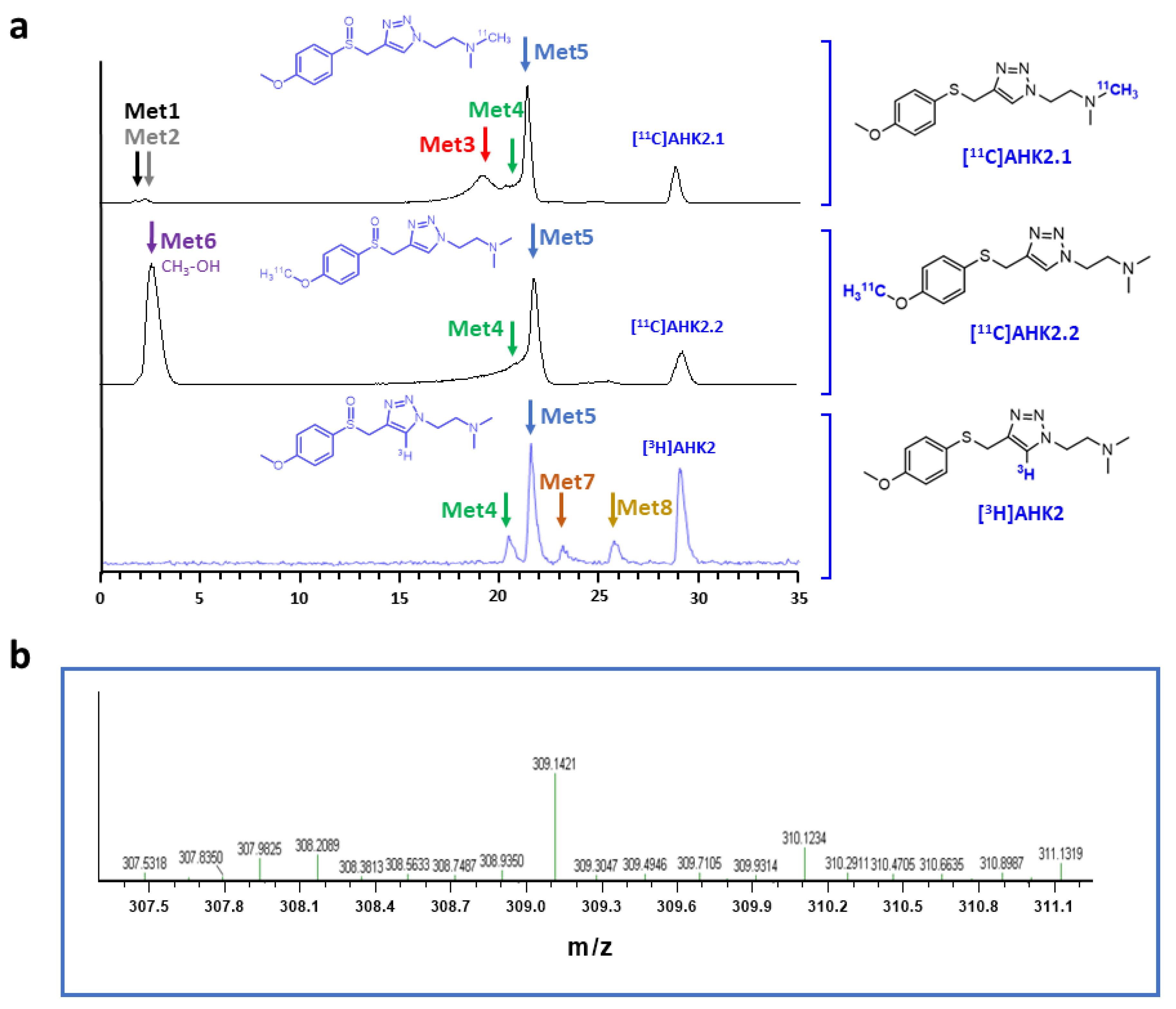
3.2. Radiochemistry—11C Labelling
3.3. Radiochemistry—3H Labelling
3.4. In Vitro Metabolism
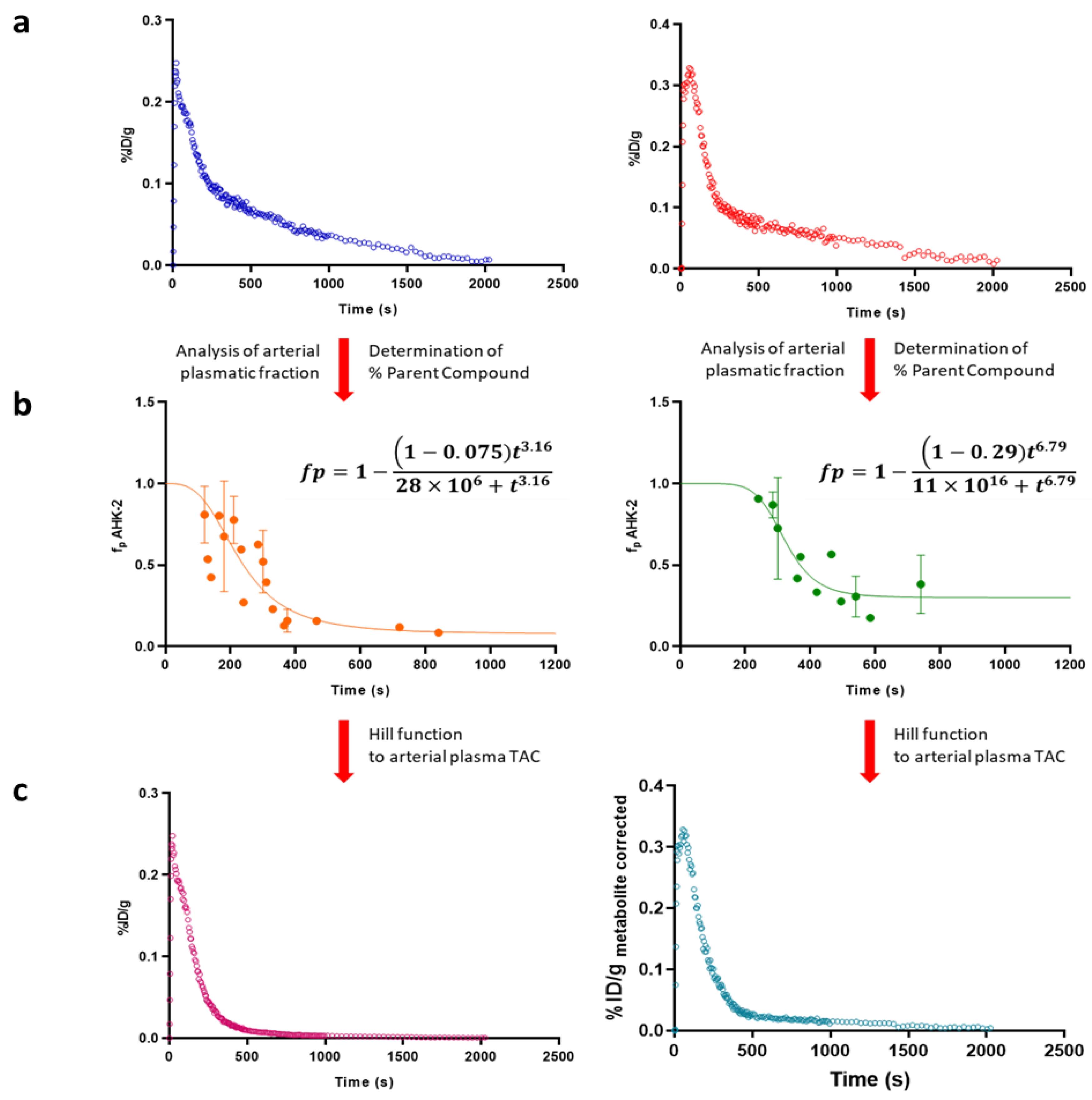
3.5. In Vivo Metabolism
3.6. Time-Activity Curves and Pharmacokinetic Analysis
3.7. In Vivo Pet Imaging
4. Discussion
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, M.H.; Baertschi, S.W.; Alsante, K.M.; Reid, D.L.; Mowery, M.D.; Shimanovich, R.; Foti, C.; Smith, W.K.; Reynolds, D.W.; Nefliu, M.; et al. In Silico Prediction of Pharmaceutical Degradation Pathways: A Benchmarking Study. Mol. Pharm. 2014, 11, 4179–4188. [Google Scholar] [CrossRef] [PubMed]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.E.; Adewumi, A.T.; Akawa, O.B.; Subair, T.I.; Okunlola, F.O.; Akinsuku, O.E.; Khan, S. Simulation Models for Prediction of Bioavailability of Medicinal Drugs-the Interface Between Experiment and Computation. AAPS PharmSciTech 2022, 23, 022–02229. [Google Scholar] [CrossRef] [PubMed]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Panse, N.; Gerk, P.M. The Caco-2 Model: Modifications and enhancements to improve efficiency and predictive performance. Int. J. Pharm. 2022, 624, 9. [Google Scholar] [CrossRef]
- Pastan, I.; Gottesman, M.M.; Ueda, K.; Lovelace, E.; Rutherford, A.V.; Willingham, M.C. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4486–4490. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.; Siramshetty, V.; Nguyễn, Ð.T.; Padilha, E.C.; Kabir, M.; Yu, K.R.; Wang, A.Q.; Zhao, T.; Itkin, M.; Shinn, P.; et al. Using in vitro ADME data for lead compound selection: An emphasis on PAMPA pH 5 permeability and oral bioavailability. Bioorg. Med. Chem. 2022, 56, 5. [Google Scholar] [CrossRef]
- Jia, L.; Liu, X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Suresh, P.S.; Srinivas, N.R.; Mullangi, R. Review of HPLC and LC-MS/MS assays for the determination of various nonsteroidal anti-androgens used in the treatment of prostate cancer. Biomed. Chromatogr. 2018, 32, 11. [Google Scholar] [CrossRef]
- Nerella, S.G.; Singh, P.; Sanam, T.; Digwal, C.S. PET Molecular Imaging in Drug Development: The Imaging and Chemistry Perspective. Front. Med. 2022, 9, 812270. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vallejo, V.; Ugarte, A.; García-Barroso, C.; Cuadrado-Tejedor, M.; Szczupak, B.; Dopeso-Reyes, I.G.; Lanciego, J.L.; García-Osta, A.; Llop, J.; Oyarzabal, J.; et al. Pharmacokinetic investigation of sildenafil using positron emission tomography and determination of its effect on cerebrospinal fluid cGMP levels. J. Neurochem. 2016, 136, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Vallejo, V.; Martín, A.; Aginagalde, M.; San Sebastian, E.; Padro, D.; Cossío, F.P.; Llop, J. Biodistribution and metabolism of 11C-labeled Kendine 91 in mice and rats. Appl. Radiat. Isot. 2012, 70, 2545–2551. [Google Scholar] [CrossRef] [PubMed]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands–where we are and where to go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Murayama, T.; Kurebayashi, N.; Ogawa, H.; Yamazawa, T.; Oyamada, H.; Suzuki, J.; Kanemaru, K.; Oguchi, K.; Iino, M.; Sakurai, T. Genotype-phenotype correlations of malignant hyperthermia and central core disease mutations in the central region of the RYR1 channel. Hum. Mutat. 2016, 37, 1231–1241. [Google Scholar] [CrossRef]
- Lawal, T.A.; Todd, J.J.; Meilleur, K.G. Ryanodine receptor 1-related myopathies: Diagnostic and therapeutic approaches. Neurotherapeutics 2018, 15, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Aizpurua, J.M.; Miranda, J.I.; Irastorza, A.; Torres, E.; Eceiza, M.; Sagartzazu-Aizpurua, M.; Ferrón, P.; Aldanondo, G.; Lasa-Fernández, H.; Marco-Moreno, P.; et al. Discovery of a novel family of FKBP12 “reshapers” and their use as calcium modulators in skeletal muscle under nitro-oxidative stress. Eur. J. Med. Chem. 2021, 213, 113160. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef]
- Martín, A.; Szczupak, B.; Gómez-Vallejo, V.; Plaza, S.; Padró, D.; Cano, A.; Llop, J. PET Imaging of Serotoninergic Neurotransmission with [11C]DASB and [18F]altanserin after Focal Cerebral Ischemia in Rats. J. Cereb. Blood Flow Metab. 2013, 33, 1967–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Vallejo, V.; González-Esparza, M.; Llop, J. Facile and improved synthesis of [11C]Me-QNB. J. Label. Compd. Radiopharm. 2012, 55, 470–473. [Google Scholar] [CrossRef]
- Gómez-Vallejo, V.; Llop, J. Fully automated and reproducible radiosynthesis of high specific activity [11C]raclopride and [11C]Pittsburgh compound-B using the combination of two commercial synthesizers. Nucl. Med. Commun. 2011, 32, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Kopf, S.; Bourriquen, F.; Li, W.; Neumann, H.; Junge, K.; Beller, M. Recent Developments for the Deuterium and Tritium Labeling of Organic Molecules. Chem. Rev. 2022, 122, 6634–6718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Gómez-Vallejo, V.; Gaja, V.; Gona, K.B.; Llop, J. Nitrogen-13: Historical review and future perspectives. J. Label. Comp. Radiopharm 2014, 57, 244–254. [Google Scholar] [CrossRef]
- Itsenko, O.; Goméz-Vallejo, V.; Llop, J.; Koziorowski, J. On 11C Chemistry Reviews—Surveying and Filling the Gaps. Curr. Org. Chem. 2013, 17, 2067–2096. [Google Scholar] [CrossRef]
- Pichler, V.; Berroterán-Infante, N.; Philippe, C.; Vraka, C.; Klebermass, E.M.; Balber, T.; Pfaff, S.; Nics, L.; Mitterhauser, M.; Wadsak, W. An Overview of PET Radiochemistry, Part 1: The Covalent Labels (18)F, (11)C, and (13)N. J. Nucl. Med. 2018, 59, 1350–1354. [Google Scholar] [CrossRef] [Green Version]
- Zanotti-Fregonara, P.; Leroy, C.; Roumenov, D.; Trichard, C.; Martinot, J.-L.; Bottlaender, M. Kinetic analysis of [11C]befloxatone in the human brain, a selective radioligand to image monoamine oxidase A. EJNMMI Res. 2013, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Campaña, C.; Gómez-Vallejo, V.; Puigivila, M.; Martin, A.; Calvo-Fernández, T.; Moya, S.E.; Larsen, S.T.; Gispert, J.D.; Llop, J. Assessing lung inflammation after nanoparticle inhalation using 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography imaging. Mol. Imaging Biol. 2014, 16, 264–273. [Google Scholar] [CrossRef]
- Hoffman, E.J.; Huang, S.C.; Phelps, M.E. Quantitation in positron emission computed tomography: 1. Effect of object size. J. Comput. Assist. Tomogr. 1979, 3, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.M.; Ellis, J.R.; Eden, M., Jr. Analysis of emission tomographic scan data: Limitations imposed by resolution and background. J. Comput. Assist. Tomogr. 1984, 8, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Kurth, J.; Möller, A.; Förster, J.; Vollmar, B.; Krause, B.J.; Wree, A.; Stenzel, J.; Lindner, T. Continuous Blood Sampling in Small Animal Positron Emission Tomography/Computed Tomography Enables the Measurement of the Arterial Input Function. J. Vis. Exp. 2019, 8, 59701. [Google Scholar] [CrossRef] [Green Version]
- Munk, O.L.; Keiding, S.; Bass, L. A method to estimate dispersion in sampling catheters and to calculate dispersion-free blood time-activity curves. Med. Phys. 2008, 35, 3471–3481. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Parameter | Value (1 µg/Kg) | Value (5 mg/Kg) | p Value | Value * |
|---|---|---|---|---|---|
| 1 | t1/2 | 11.67 ± 3.22 | 13.60 ± 3.42 | 0.38 | 12.63 ± 3.29 |
| 2 | Cl | 1.91 ± 0.41 | 1.87 ± 0.73 | 0.71 | 1.89 ± 0.55 |
| 3 | Vz | 1996.50 ± 429.72 | 2335.03 ± 608.77 | 0.66 | 2165.76 ± 1125.06 |
| 4 | Vss | 1026.88 ± 695.33 | 1126.13 ± 443.15 | 0.75 | 1076.50 ± 808.72 |
| 5 | AUC | 53.99 ± 11.04 | 59.39 ± 19.76 | 0.6 | 56.69 ± 15.36 |
| 6 | MRT | 8.48 ± 3.76 | 8.59 ± 5.23 | 0.67 | 8.53 ± 4.30 |
| Entry | Parameter | Value (1 µg/Kg) | Value (5 mg/Kg) | p Value | Value * |
|---|---|---|---|---|---|
| 1 | t1/2 | 9.16 ± 3.78 | 12.97 ± 11.06 | 0.6 | 11.34 ± 8.44 |
| 2 | Cl | 4.23 ± 3.58 | 1.57 ± 0.69 | 0.29 | 2.71 ± 2.55 |
| 3 | Vz | 2728.14 ± 1252.35 | 1624.28 ± 1299.35 | 0.31 | 2097.36 ± 1309.61 |
| 4 | AUC | 6.68 ± 4.35 | 13.35 ± 5.08 | 0.32 | 10.49 ± 5.65 |
| 5 | MRT | 8.51 ± 2.69 | 9.36 ± 8.51 | 0.88 | 9.00 ± 6.93 |
| 6 | F | 0.185 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passannante, R.; Gómez-Vallejo, V.; Sagartzazu-Aizpurua, M.; Vignau Arsuaga, L.; Marco-Moreno, P.; Aldanondo, G.; Vallejo-Illarramendi, A.; Aguiar, P.; Cossío, U.; Martín, A.; et al. Pharmacokinetic Evaluation of New Drugs Using a Multi-Labelling Approach and PET Imaging: Application to a Drug Candidate with Potential Application in Neuromuscular Disorders. Biomedicines 2023, 11, 253. https://doi.org/10.3390/biomedicines11020253
Passannante R, Gómez-Vallejo V, Sagartzazu-Aizpurua M, Vignau Arsuaga L, Marco-Moreno P, Aldanondo G, Vallejo-Illarramendi A, Aguiar P, Cossío U, Martín A, et al. Pharmacokinetic Evaluation of New Drugs Using a Multi-Labelling Approach and PET Imaging: Application to a Drug Candidate with Potential Application in Neuromuscular Disorders. Biomedicines. 2023; 11(2):253. https://doi.org/10.3390/biomedicines11020253
Chicago/Turabian StylePassannante, Rossana, Vanessa Gómez-Vallejo, Maialen Sagartzazu-Aizpurua, Laura Vignau Arsuaga, Pablo Marco-Moreno, Garazi Aldanondo, Ainara Vallejo-Illarramendi, Pablo Aguiar, Unai Cossío, Abraham Martín, and et al. 2023. "Pharmacokinetic Evaluation of New Drugs Using a Multi-Labelling Approach and PET Imaging: Application to a Drug Candidate with Potential Application in Neuromuscular Disorders" Biomedicines 11, no. 2: 253. https://doi.org/10.3390/biomedicines11020253
APA StylePassannante, R., Gómez-Vallejo, V., Sagartzazu-Aizpurua, M., Vignau Arsuaga, L., Marco-Moreno, P., Aldanondo, G., Vallejo-Illarramendi, A., Aguiar, P., Cossío, U., Martín, A., Bergare, J., Kingston, L., Elmore, C. S., Morcillo, M. A., Ferrón, P., Aizpurua, J. M., & Llop, J. (2023). Pharmacokinetic Evaluation of New Drugs Using a Multi-Labelling Approach and PET Imaging: Application to a Drug Candidate with Potential Application in Neuromuscular Disorders. Biomedicines, 11(2), 253. https://doi.org/10.3390/biomedicines11020253