
Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Publicly Available Transcriptomic and Proteomic Data
2.2. Functional Annotation of Reproducible Genes in BTBR Mice
2.3. Association of a Genetic Background and Gene Expression
2.4. Reanalysis of the Published RNA-Seq Data from the Cortex and Hippocampus of BTBR Mice
2.5. A Comparison between BTBR-Related Genes and Genes Associated with Human Autism or SCZ
3. Results
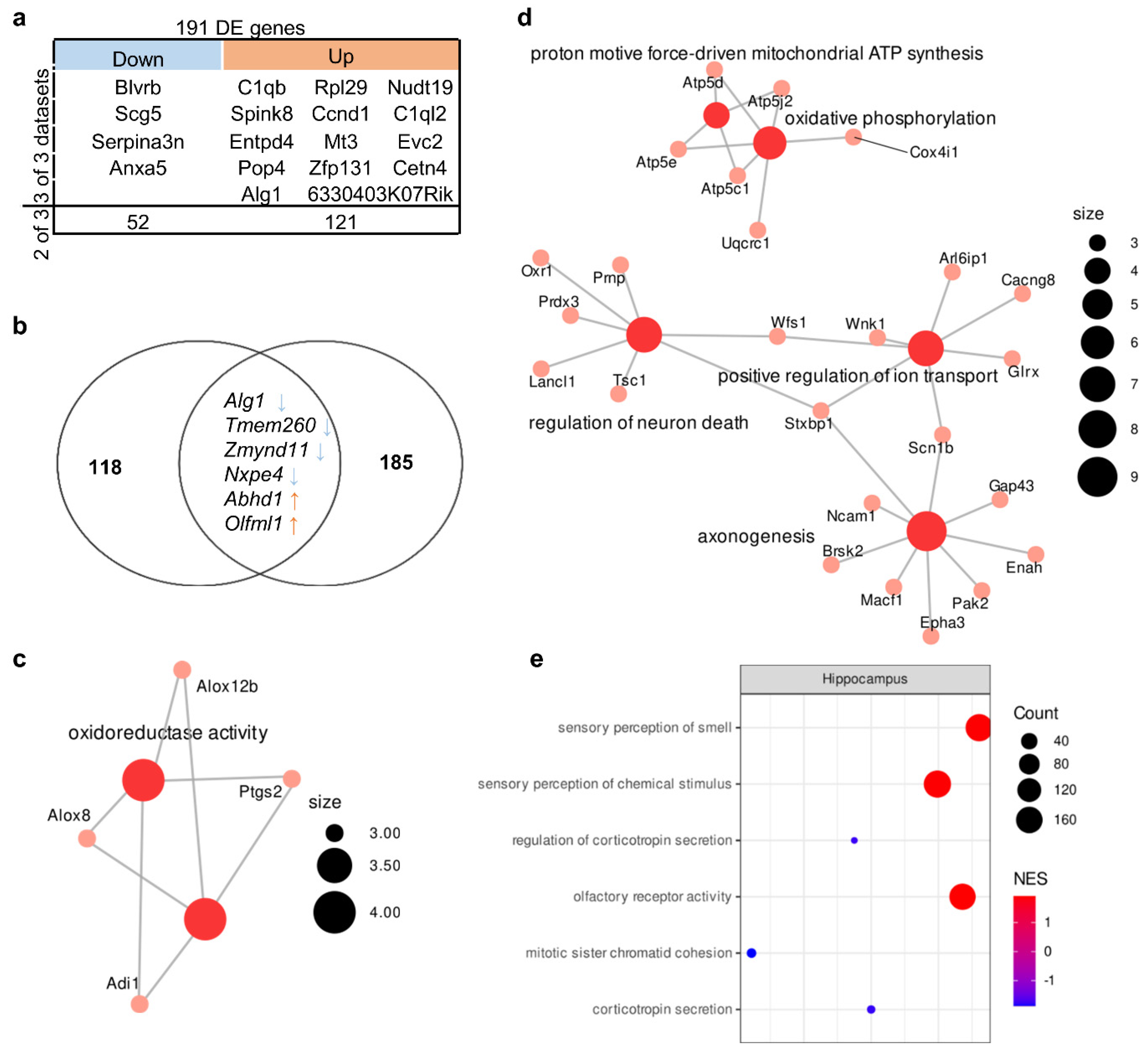
3.1. Transcriptome Aberrations in the Hippocampus of BTBR Mice
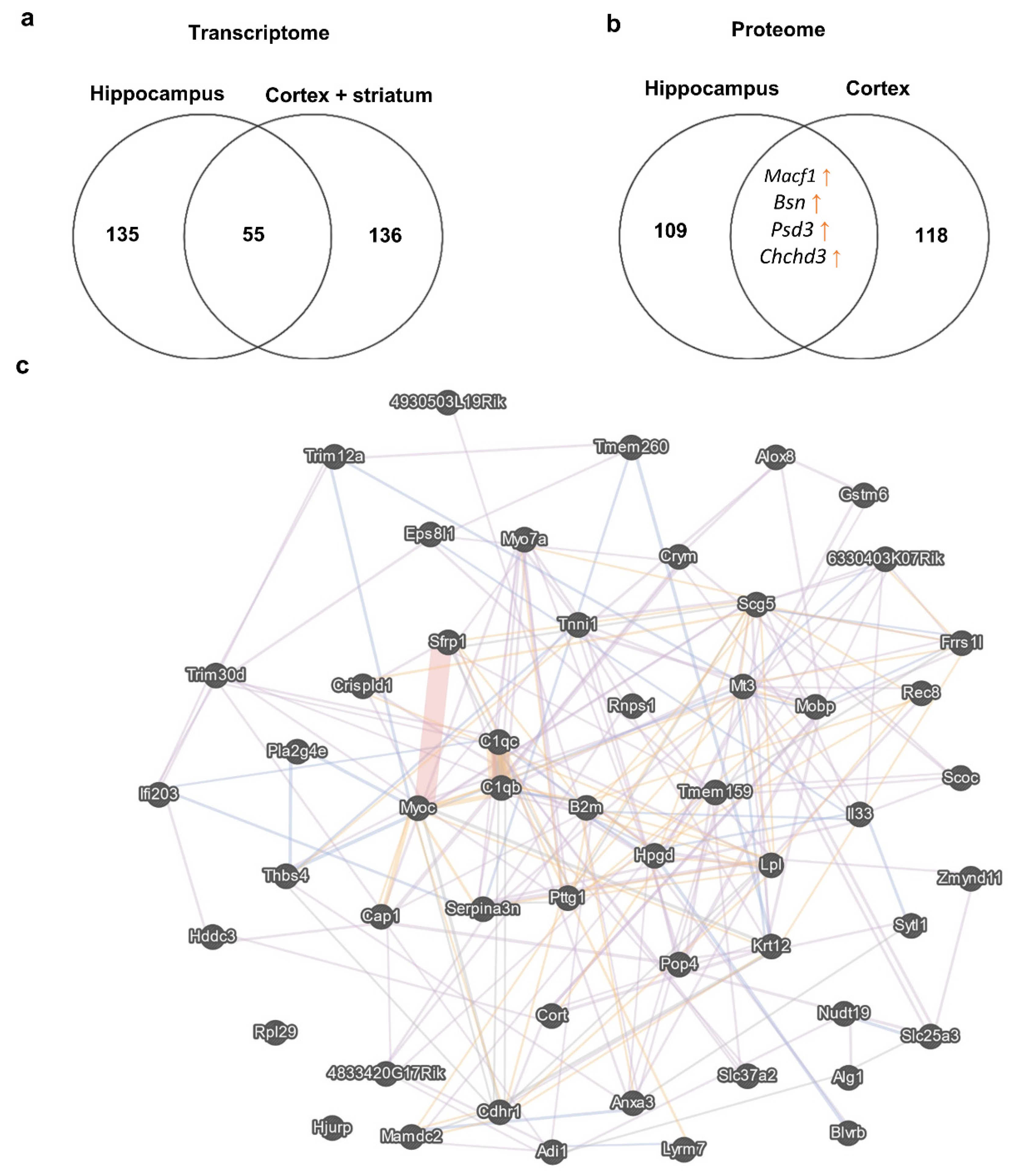
3.2. Transcriptome Alterations in the Corticostriatal Area of BTBR Mice
3.3. Genetic Characteristics of BTBR Mice
3.4. The Comparison between DEGs from Postmortem ASD or SCZ Human Tissue Samples and DEGs from BTBR Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A Review of Worldwide Prevalence Estimates Since 2014. Brain Sci. 2020, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, L.; Chen, H.; Fang, Y.; Zhang, T.; Yin, X.; Man, J.; Yang, X.; Lu, M. Global, Regional and National Burden of Autism Spectrum Disorder from 1990 to 2019: Results from the Global Burden of Disease Study 2019. Epidemiol. Psychiatr. Sci. 2022, 31, e33. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenco, S.; Weinberger, D.R. The Neurodevelopmental Hypothesis of Schizophrenia: Following a Trail of Evidence from Cradle to Grave. Dev. Psychopathol. 2000, 12, 501–527. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Folsom, T.D. The Neurodevelopmental Hypothesis of Schizophrenia, Revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Insel, T.R. Rethinking Schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.M.; Bhavsar, V.; Tripoli, G.; Howes, O. 30 Years on: How the Neurodevelopmental Hypothesis of Schizophrenia Morphed into the Developmental Risk Factor Model of Psychosis. Schizophr. Bull. 2017, 43, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Egerton, A.; Grace, A.A.; Stone, J.; Bossong, M.G.; Sand, M.; McGuire, P. Glutamate in Schizophrenia: Neurodevelopmental Perspectives and Drug Development. Schizophr. Res. 2020, 223, 59–70. [Google Scholar] [CrossRef]
- King, B.H.; Lord, C. Is Schizophrenia on the Autism Spectrum? Brain Res. 2011, 1380, 34–41. [Google Scholar] [CrossRef]
- de Lacy, N.; King, B.H. Revisiting the Relationship between Autism and Schizophrenia: Toward an Integrated Neurobiology. Annu. Rev. Clin. Psychol. 2013, 9, 555–587. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; O’Donovan, M.C. Schizophrenia and the Neurodevelopmental Continuum:Evidence from Genomics. World Psychiatry 2017, 16, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A Copy Number Variation Morbidity Map of Developmental Delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative Burden of Large CNVs on a Range of Neurodevelopmental Phenotypes. PLoS Genet. 2011, 7, e1002334. [Google Scholar] [CrossRef]
- Kirov, G.; Rees, E.; Walters, J.T.R.; Escott-Price, V.; Georgieva, L.; Richards, A.L.; Chambert, K.D.; Davies, G.; Legge, S.E.; Moran, J.L.; et al. The Penetrance of Copy Number Variations for Schizophrenia and Developmental Delay. Biol. Psychiatry 2014, 75, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Zheng, P.; Zou, X. Association between Schizophrenia and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Autism Res. Off. J. Int. Soc. Autism Res. 2018, 11, 1110–1119. [Google Scholar] [CrossRef]
- Lai, M.-C.; Kassee, C.; Besney, R.; Bonato, S.; Hull, L.; Mandy, W.; Szatmari, P.; Ameis, S.H. Prevalence of Co-Occurring Mental Health Diagnoses in the Autism Population: A Systematic Review and Meta-Analysis. Lancet. Psychiatry 2019, 6, 819–829. [Google Scholar] [CrossRef]
- Jutla, A.; Foss-Feig, J.; Veenstra-VanderWeele, J. Autism Spectrum Disorder and Schizophrenia: An Updated Conceptual Review. Autism Res. Off. J. Int. Soc. Autism Res. 2022, 15, 384–412. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Warrier, V.; Zhang, X.; Reed, P.; Havdahl, A.; Moore, T.M.; Cliquet, F.; Leblond, C.S.; Rolland, T.; Rosengren, A.; Rowitch, D.H.; et al. Genetic Correlates of Phenotypic Heterogeneity in Autism. Nat. Genet. 2022, 54, 1293–1304. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Qiu, Y.; Li, Y.; Cong, X. Genetics of Autism Spectrum Disorder: An Umbrella Review of Systematic Reviews and Meta-Analyses. Transl. Psychiatry 2022, 12, 249. [Google Scholar] [CrossRef] [PubMed]
- Mahony, C.; O’Ryan, C. Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data. Int. J. Mol. Sci. 2021, 22, 10757. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.E.; Panitch, R.; West, A.B.; Arking, D.E. Transcriptome Analysis of Cortical Tissue Reveals Shared Sets of Downregulated Genes in Autism and Schizophrenia. Transl. Psychiatry 2016, 6, e817. [Google Scholar] [CrossRef] [Green Version]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-Wide Isoform-Level Dysregulation in ASD, Schizophrenia, and Bipolar Disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [Green Version]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared Molecular Neuropathology across Major Psychiatric Disorders Parallels Polygenic Overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Cai, J.J.; Ji, G.; Sham, P.C. Commonality in Dysregulated Expression of Gene Sets in Cortical Brains of Individuals with Autism, Schizophrenia, and Bipolar Disorder. Transl. Psychiatry 2019, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Nomura, J.; Mardo, M.; Takumi, T. Molecular Signatures from Multi-Omics of Autism Spectrum Disorders and Schizophrenia. J. Neurochem. 2021, 159, 647–659. [Google Scholar] [CrossRef]
- Gao, R.; Penzes, P. Common Mechanisms of Excitatory and Inhibitory Imbalance in Schizophrenia and Autism Spectrum Disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef]
- Scattoni, M.L.; Martire, A.; Cartocci, G.; Ferrante, A.; Ricceri, L. Reduced Social Interaction, Behavioural Flexibility and BDNF Signalling in the BTBR T+tf/J Strain, a Mouse Model of Autism. Behav. Brain Res. 2013, 251, 35–40. [Google Scholar] [CrossRef]
- Ellegood, J.; Crawley, J.N. Behavioral and Neuroanatomical Phenotypes in Mouse Models of Autism. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshetnikov, V.V.; Ayriyants, K.A.; Ryabushkina, Y.A.; Sozonov, N.G.; Bondar, N.P. Sex-Specific Behavioral and Structural Alterations Caused by Early-Life Stress in C57BL/6 and BTBR Mice. Behav. Brain Res. 2021, 414, 113489. [Google Scholar] [CrossRef] [PubMed]
- Meyza, K.Z.; Blanchard, D.C. The BTBR Mouse Model of Idiopathic Autism—Current View on Mechanisms. Neurosci. Biobehav. Rev. 2017, 76, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, S.; Hirota, J.-N.; Ishii, C.; Iwasaki, H.; Sano, Y.; Furuichi, T. Comprehensive Profiling of Gene Expression in the Cerebral Cortex and Striatum of BTBRTF/ArtRbrc Mice Compared to C57BL/6J Mice. Front. Cell. Neurosci. 2020, 14, 595607. [Google Scholar] [CrossRef]
- Bove, M.; Ike, K.; Eldering, A.; Buwalda, B.; de Boer, S.F.; Morgese, M.G.; Schiavone, S.; Cuomo, V.; Trabace, L.; Kas, M.J.H. The Visible Burrow System: A Behavioral Paradigm to Assess Sociability and Social Withdrawal in BTBR and C57BL/6J Mice Strains. Behav. Brain Res. 2018, 344, 9–19. [Google Scholar] [CrossRef]
- Gogolla, N.; Takesian, A.E.; Feng, G.; Fagiolini, M.; Hensch, T.K. Sensory Integration in Mouse Insular Cortex Reflects GABA Circuit Maturation. Neuron 2014, 83, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Clapcote, S.J.; Roder, J.C. Deletion Polymorphism of Disc1 Is Common to All 129 Mouse Substrains: Implications for Gene-Targeting Studies of Brain Function. Genetics 2006, 173, 2407–2410. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, S.; Del Vecchio, G.; Gioiosa, S.; Flati, T.; Castrignano, T.; Legnini, I.; Licursi, V.; Ricceri, L.; Scattoni, M.L.; Rinaldi, A.; et al. Differential Expression of Hippocampal Circular RNAs in the BTBR Mouse Model for Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2301–2313. [Google Scholar] [CrossRef]
- Provenzano, G.; Corradi, Z.; Monsorno, K.; Fedrizzi, T.; Ricceri, L.; Scattoni, M.L.; Bozzi, Y. Comparative Gene Expression Analysis of Two Mouse Models of Autism: Transcriptome Profiling of the BTBR and En2−/− Hippocampus. Front. Neurosci. 2016, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Daimon, C.M.; Jasien, J.M.; Wood, W.H.; Zhang, Y.; Becker, K.G.; Silverman, J.L.; Crawley, J.N.; Martin, B.; Maudsley, S. Hippocampal Transcriptomic and Proteomic Alterations in the BTBR Mouse Model of Autism Spectrum Disorder. Front. Physiol. 2015, 6, 324. [Google Scholar] [CrossRef]
- Wang, M.; He, J.; Zhou, Y.; Lv, N.; Zhao, M.; Wei, H.; Li, R. Integrated Analysis of MiRNA and MRNA Expression Profiles in the Brains of BTBR Mice. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2020, 80, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Oron, O.; Getselter, D.; Shohat, S.; Reuveni, E.; Lukic, I.; Shifman, S.; Elliott, E. Gene Network Analysis Reveals a Role for Striatal Glutamatergic Receptors in Dysregulated Risk-Assessment Behavior of Autism Mouse Models. Transl. Psychiatry 2019, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Ma, Y.; Liu, J.; Ding, C.; Hu, F.; Yu, L. Proteomic Analysis of Cortical Brain Tissue from the BTBR Mouse Model of Autism: Evidence for Changes in STOP and Myelin-Related Proteins. Neuroscience 2016, 312, 26–34. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshetnikov, V.V.; Kisaretova, P.E.; Ershov, N.I.; Shulyupova, A.S.; Oshchepkov, D.Y.; Klimova, N.V.; Ivanchihina, A.V.; Merkulova, T.I.; Bondar, N.P. Genes Associated with Cognitive Performance in the Morris Water Maze: An RNA-Seq Study. Sci. Rep. 2020, 10, 22078. [Google Scholar] [CrossRef]
- Reshetnikov, V.V.; Kisaretova, P.E.; Ershov, N.I.; Merkulova, T.I.; Bondar, N.P. Social Defeat Stress in Adult Mice Causes Alterations in Gene Expression, Alternative Splicing, and the Epigenetic Landscape of H3K4me3 in the Prefrontal Cortex: An Impact of Early-Life Stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 106, 110068. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Forés-Martos, J.; Catalá-López, F.; Sánchez-Valle, J.; Ibáñez, K.; Tejero, H.; Palma-Gudiel, H.; Climent, J.; Pancaldi, V.; Fañanás, L.; Arango, C.; et al. Transcriptomic Metaanalyses of Autistic Brains Reveals Shared Gene Expression and Biological Pathway Abnormalities with Cancer. Mol. Autism 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.R.; Petralia, M.C.; Ciurleo, R.; Bramanti, A.; Fagone, P.; Shahjaman, M.; Wu, L.; Sun, Y.; Turanli, B.; Arga, K.Y.; et al. Comprehensive Analysis of RNA-Seq Gene Expression Profiling of Brain Transcriptomes Reveals Novel Genes, Regulators, and Pathways in Autism Spectrum Disorder. Brain Sci. 2020, 10, 747. [Google Scholar] [CrossRef]
- Manchia, M.; Piras, I.S.; Huentelman, M.J.; Pinna, F.; Zai, C.C.; Kennedy, J.L.; Carpiniello, B. Pattern of Gene Expression in Different Stages of Schizophrenia: Down-Regulation of NPTX2 Gene Revealed by a Meta-Analysis of Microarray Datasets. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 1054–1063. [Google Scholar] [CrossRef]
- Qin, W.; Liu, C.; Sodhi, M.; Lu, H. Meta-Analysis of Sex Differences in Gene Expression in Schizophrenia. BMC Syst. Biol. 2016, 10 (Suppl. S1), 9. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Venkataraman, L.; Chen, S.; Fu, H. Function of WFS1 and WFS2 in the Central Nervous System: Implications for Wolfram Syndrome and Alzheimer’s Disease. Neurosci. Biobehav. Rev. 2020, 118, 775–783. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Cao, X.; Yin, F.; Wu, T.; Stauber, T.; Peng, J. West Syndrome Caused by a Chloride/Proton Exchange-Uncoupling CLCN6 Mutation Related to Autophagic-Lysosomal Dysfunction. Mol. Neurobiol. 2021, 58, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, A.T.; Wang, M.; Hauberg, M.E.; Fullard, J.F.; Kozlenkov, A.; Keenan, A.; Hurd, Y.L.; Dracheva, S.; Casaccia, P.; Roussos, P.; et al. Brain Cell Type Specific Gene Expression and Co-Expression Network Architectures. Sci. Rep. 2018, 8, 8868. [Google Scholar] [CrossRef] [Green Version]
- Altin, J.G.; Sloan, E.K. The Role of CD45 and CD45-Associated Molecules in T Cell Activation. Immunol. Cell Biol. 1997, 75, 430–445. [Google Scholar] [CrossRef]
- Marsango, S.; Barki, N.; Jenkins, L.; Tobin, A.B.; Milligan, G. Therapeutic Validation of an Orphan G Protein-Coupled Receptor: The Case of GPR84. Br. J. Pharmacol. 2022, 179, 3529–3541. [Google Scholar] [CrossRef]
- Pappaianni, E.; Siugzdaite, R.; Vettori, S.; Venuti, P.; Job, R.; Grecucci, A. Three Shades of Grey: Detecting Brain Abnormalities in Children with Autism Using Source-, Voxel- and Surface-Based Morphometry. Eur. J. Neurosci. 2018, 47, 690–700. [Google Scholar] [CrossRef]
- Crucitti, J.; Hyde, C.; Enticott, P.G.; Stokes, M.A. A Systematic Review of Frontal Lobe Volume in Autism Spectrum Disorder Revealing Distinct Trajectories. J. Integr. Neurosci. 2022, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Hardan, A.Y.; Muddasani, S.; Vemulapalli, M.; Keshavan, M.S.; Minshew, N.J. An MRI Study of Increased Cortical Thickness in Autism. Am. J. Psychiatry 2006, 163, 1290–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, R.; Mostofsky, S.H. Neuroimaging Endophenotypes in Autism Spectrum Disorder. CNS Spectr. 2015, 20, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Libero, L.E.; Burge, W.K.; Deshpande, H.D.; Pestilli, F.; Kana, R.K. White Matter Diffusion of Major Fiber Tracts Implicated in Autism Spectrum Disorder. Brain Connect. 2016, 6, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Lucibello, S.; Bertè, G.; Verdolotti, T.; Lucignani, M.; Napolitano, A.; D’Abronzo, R.; Cicala, M.G.; Pede, E.; Chieffo, D.; Mariotti, P.; et al. Cortical Thickness and Clinical Findings in Prescholar Children with Autism Spectrum Disorder. Front. Neurosci. 2021, 15, 776860. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.P.A.; Basson, M.A. The Neuroanatomy of Autism—A Developmental Perspective. J. Anat. 2017, 230, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Sparks, B.F.; Friedman, S.D.; Shaw, D.W.; Aylward, E.H.; Echelard, D.; Artru, A.A.; Maravilla, K.R.; Giedd, J.N.; Munson, J.; Dawson, G.; et al. Brain Structural Abnormalities in Young Children with Autism Spectrum Disorder. Neurology 2002, 59, 184–192. [Google Scholar] [CrossRef]
- Schumann, C.M.; Hamstra, J.; Goodlin-Jones, B.L.; Lotspeich, L.J.; Kwon, H.; Buonocore, M.H.; Lammers, C.R.; Reiss, A.L.; Amaral, D.G. The Amygdala Is Enlarged in Children but not Adolescents with Autism; the Hippocampus Is Enlarged at All Ages. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 6392–6401. [Google Scholar] [CrossRef] [Green Version]
- Rojas, D.C.; Peterson, E.; Winterrowd, E.; Reite, M.L.; Rogers, S.J.; Tregellas, J.R. Regional Gray Matter Volumetric Changes in Autism Associated with Social and Repetitive Behavior Symptoms. BMC Psychiatry 2006, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Groen, W.; Teluij, M.; Buitelaar, J.; Tendolkar, I. Amygdala and Hippocampus Enlargement during Adolescence in Autism. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 552–560. [Google Scholar] [CrossRef]
- Hasan, K.M.; Walimuni, I.S.; Frye, R.E. Global Cerebral and Regional Multimodal Neuroimaging Markers of the Neurobiology of Autism: Development and Cognition. J. Child Neurol. 2013, 28, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Wang, D.; Tao, F.; Wang, Y. Changes in the Development of Subcortical Structures in Autism Spectrum Disorder. Neuroreport 2019, 30, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, V.P.; Iosif, A.-M.; Libero, L.; Heath, B.; Rogers, S.J.; Ferrer, E.; Nordahl, C.; Ghetti, S.; Amaral, D.; Solomon, M. Understanding Hippocampal Development in Young Children with Autism Spectrum Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2020, 59, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, M.-H.; Li, G.; Wu, D.; Lian, C.; Sun, Q.; Rushmore, R.J.; Wang, L. Volumetric Analysis of Amygdala and Hippocampal Subfields for Infants with Autism. J. Autism Dev. Disord. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.; Greimel, E.; Kliemann, D.; Koerte, I.K.; Schulte-Körne, G.; Reuter, M.; Wachinger, C. Increased Hippocampal Shape Asymmetry and Volumetric Ventricular Asymmetry in Autism Spectrum Disorder. NeuroImage. Clin. 2020, 26, 102207. [Google Scholar] [CrossRef]
- Qin, B.; Wang, L.; Cai, J.; Li, T.; Zhang, Y. Functional Brain Networks in Preschool Children with Autism Spectrum Disorders. Front. Psychiatry 2022, 13, 896388. [Google Scholar] [CrossRef]
- Qiu, A.; Adler, M.; Crocetti, D.; Miller, M.I.; Mostofsky, S.H. Basal Ganglia Shapes Predict Social, Communication, and Motor Dysfunctions in Boys with Autism Spectrum Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 539–551. [Google Scholar] [CrossRef]
- Schuetze, M.; Park, M.T.M.; Cho, I.Y.; MacMaster, F.P.; Chakravarty, M.M.; Bray, S.L. Morphological Alterations in the Thalamus, Striatum, and Pallidum in Autism Spectrum Disorder. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 2627–2637. [Google Scholar] [CrossRef] [Green Version]
- Voelbel, G.T.; Bates, M.E.; Buckman, J.F.; Pandina, G.; Hendren, R.L. Caudate Nucleus Volume and Cognitive Performance: Are They Related in Childhood Psychopathology? Biol. Psychiatry 2006, 60, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Langen, M.; Durston, S.; Staal, W.G.; Palmen, S.J.M.C.; van Engeland, H. Caudate Nucleus Is Enlarged in High-Functioning Medication-Naive Subjects with Autism. Biol. Psychiatry 2007, 62, 262–266. [Google Scholar] [CrossRef]
- Estes, A.; Shaw, D.W.W.; Sparks, B.F.; Friedman, S.; Giedd, J.N.; Dawson, G.; Bryan, M.; Dager, S.R. Basal Ganglia Morphometry and Repetitive Behavior in Young Children with Autism Spectrum Disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2011, 4, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, D.; Leung, R.C.; Vogan, V.M.; Lee, W.; Trelle, S.; Lin, S.; Cassel, D.B.; Chakravarty, M.M.; Lerch, J.P.; Anagnostou, E.; et al. The Autism Puzzle: Diffuse but Not Pervasive Neuroanatomical Abnormalities in Children with ASD. NeuroImage Clin. 2015, 8, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, D.; Anagnostou, E.; Arango, C.; Auzias, G.; Behrmann, M.; Busatto, G.F.; Calderoni, S.; Daly, E.; Deruelle, C.; Di Martino, A.; et al. Cortical and Subcortical Brain Morphometry Differences between Patients with Autism Spectrum Disorder and Healthy Individuals across the Lifespan: Results from the ENIGMA ASD Working Group. Am. J. Psychiatry 2018, 175, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.-Y.; Liu, F.-C. Pathological Alterations in Striatal Compartments in the Human Brain of Autism Spectrum Disorder. Mol. Brain 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, A.; Kelly, C.; Grzadzinski, R.; Zuo, X.-N.; Mennes, M.; Mairena, M.A.; Lord, C.; Castellanos, F.X.; Milham, M.P. Aberrant Striatal Functional Connectivity in Children with Autism. Biol. Psychiatry 2011, 69, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janouschek, H.; Chase, H.W.; Sharkey, R.J.; Peterson, Z.J.; Camilleri, J.A.; Abel, T.; Eickhoff, S.B.; Nickl-Jockschat, T. The Functional Neural Architecture of Dysfunctional Reward Processing in Autism. NeuroImage Clin. 2021, 31, 102700. [Google Scholar] [CrossRef]
- Rubenstein, J.L.R. Annual Research Review: Development of the Cerebral Cortex: Implications for Neurodevelopmental Disorders. J. Child Psychol. Psychiatry 2011, 52, 339–355. [Google Scholar] [CrossRef]
- Meechan, D.W.; Maynard, T.M.; Tucker, E.S.; Fernandez, A.; Karpinski, B.A.; Rothblat, L.A.; LaMantia, A.-S. Modeling a Model: Mouse Genetics, 22q11.2 Deletion Syndrome, and Disorders of Cortical Circuit Development. Prog. Neurobiol. 2015, 130, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Fenlon, L.R.; Liu, S.; Gobius, I.; Kurniawan, N.D.; Murphy, S.; Moldrich, R.X.; Richards, L.J. Formation of Functional Areas in the Cerebral Cortex Is Disrupted in a Mouse Model of Autism Spectrum Disorder. Neural Dev. 2015, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Takumi, T.; Tamada, K.; Hatanaka, F.; Nakai, N.; Bolton, P.F. Behavioral Neuroscience of Autism. Neurosci. Biobehav. Rev. 2020, 110, 60–76. [Google Scholar] [CrossRef]
- Schmeisser, M.J. Translational Neurobiology in Shank Mutant Mice--Model Systems for Neuropsychiatric Disorders. Ann. Anat. Anat. Anz. Off. Organ Anat. Ges. 2015, 200, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, C.; Yau, S.-Y.; Majaess, N.; Vetrici, M.; Gil-Mohapel, J.; Christie, B.R. Hippocampal Dysfunction and Cognitive Impairment in Fragile-X Syndrome. Neurosci. Biobehav. Rev. 2016, 68, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Fuccillo, M. V Striatal Circuits as a Common Node for Autism Pathophysiology. Front. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, S.; De Zeeuw, C.I.; Boeckers, T.M.; Schmeisser, M.J. Cerebellar and Striatal Pathologies in Mouse Models of Autism Spectrum Disorder. Adv. Anat. Embryol. Cell Biol. 2017, 224, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Thabault, M.; Turpin, V.; Maisterrena, A.; Jaber, M.; Egloff, M.; Galvan, L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. Int. J. Mol. Sci. 2022, 23, 2294. [Google Scholar] [CrossRef]
- Tsurugizawa, T. Translational Magnetic Resonance Imaging in Autism Spectrum Disorder from the Mouse Model to Human. Front. Neurosci. 2022, 16, 872036. [Google Scholar] [CrossRef]
- Meyza, K.Z.; Defensor, E.B.; Jensen, A.L.; Corley, M.J.; Pearson, B.L.; Pobbe, R.L.H.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. The BTBR T+ Tf/J Mouse Model for Autism Spectrum Disorders-in Search of Biomarkers. Behav. Brain Res. 2013, 251, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Dodero, L.; Damiano, M.; Galbusera, A.; Bifone, A.; Tsaftsaris, S.A.; Scattoni, M.L.; Gozzi, A. Neuroimaging Evidence of Major Morpho-Anatomical and Functional Abnormalities in the BTBR T+TF/J Mouse Model of Autism. PLoS ONE 2013, 8, e76655. [Google Scholar] [CrossRef] [Green Version]
- Faraji, J.; Karimi, M.; Lawrence, C.; Mohajerani, M.H.; Metz, G.A.S. Non-Diagnostic Symptoms in a Mouse Model of Autism in Relation to Neuroanatomy: The BTBR Strain Reinvestigated. Transl. Psychiatry 2018, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Ellegood, J.; Babineau, B.A.; Henkelman, R.M.; Lerch, J.P.; Crawley, J.N. Neuroanatomical Analysis of the BTBR Mouse Model of Autism Using Magnetic Resonance Imaging and Diffusion Tensor Imaging. Neuroimage 2013, 70, 288–300. [Google Scholar] [CrossRef]
- Pagani, M.; Damiano, M.; Galbusera, A.; Tsaftaris, S.A.; Gozzi, A. Semi-Automated Registration-Based Anatomical Labelling, Voxel Based Morphometry and Cortical Thickness Mapping of the Mouse Brain. J. Neurosci. Methods 2016, 267, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Sforazzini, F.; Bertero, A.; Dodero, L.; David, G.; Galbusera, A.; Scattoni, M.L.; Pasqualetti, M.; Gozzi, A. Altered Functional Connectivity Networks in Acallosal and Socially Impaired BTBR Mice. Brain Struct. Funct. 2016, 221, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Squillace, M.; Dodero, L.; Federici, M.; Migliarini, S.; Errico, F.; Napolitano, F.; Krashia, P.; Di Maio, A.; Galbusera, A.; Bifone, A.; et al. Dysfunctional Dopaminergic Neurotransmission in Asocial BTBR Mice. Transl. Psychiatry 2014, 4, e427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, V.M.; Gupta, D.; Neu, N.; Cotroneo, A.; Boulay, C.B.; Seegal, R.F. Novel Inter-Hemispheric White Matter Connectivity in the BTBR Mouse Model of Autism. Brain Res. 2013, 1513, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbikay, M.; Seidah, N.G.; Chrétien, M. Neuroendocrine Secretory Protein 7B2: Structure, Expression and Functions. Biochem. J. 2001, 357, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Chaplot, K.; Jarvela, T.S.; Lindberg, I. Secreted Chaperones in Neurodegeneration. Front. Aging Neurosci. 2020, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Kimble, A.; Peng, B.; Zhu, P.; Pintar, J.E.; Steiner, D.F.; Lindberg, I. Mortality in 7B2 Null Mice Can Be Rescued by Adrenalectomy: Involvement of Dopamine in ACTH Hypersecretion. Proc. Natl. Acad. Sci. USA 2002, 99, 3087–3092. [Google Scholar] [CrossRef] [Green Version]
- Peinado, J.R.; Laurent, V.; Lee, S.-N.; Peng, B.W.; Pintar, J.E.; Steiner, D.F.; Lindberg, I. Strain-Dependent Influences on the Hypothalamo-Pituitary-Adrenal Axis Profoundly Affect the 7B2 and PC2 Null Phenotypes. Endocrinology 2005, 146, 3438–3444. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-N.; Lindberg, I. 7B2 Prevents Unfolding and Aggregation of Prohormone Convertase 2. Endocrinology 2008, 149, 4116–4127. [Google Scholar] [CrossRef] [Green Version]
- Helwig, M.; Hoshino, A.; Berridge, C.; Lee, S.-N.; Lorenzen, N.; Otzen, D.E.; Eriksen, J.L.; Lindberg, I. The Neuroendocrine Protein 7B2 Suppresses the Aggregation of Neurodegenerative Disease-Related Proteins. J. Biol. Chem. 2013, 288, 1114–1124. [Google Scholar] [CrossRef]
- Iguchi, H.; Chan, J.S.; Seidah, N.G.; Chrétien, M. Evidence for a Novel Pituitary Protein (7B2) in Human Brain, Cerebrospinal Fluid and Plasma: Brain Concentrations in Controls and Patients with Alzheimer’s Disease. Peptides 1987, 8, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Winsky-Sommerer, R.; Grouselle, D.; Rougeot, C.; Laurent, V.; David, J.-P.; Delacourte, A.; Dournaud, P.; Seidah, N.G.; Lindberg, I.; Trottier, S.; et al. The Proprotein Convertase PC2 Is Involved in the Maturation of Prosomatostatin to Somatostatin-14 but Not in the Somatostatin Deficit in Alzheimer’s Disease. Neuroscience 2003, 122, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Kato, C.; Tochigi, M.; Koishi, S.; Kawakubo, Y.; Yamamoto, K.; Matsumoto, H.; Hashimoto, O.; Kim, S.-Y.; Watanabe, K.; Kano, Y.; et al. Association Study of the Commonly Recognized Breakpoints in Chromosome 15q11-Q13 in Japanese Autistic Patients. Psychiatr. Genet. 2008, 18, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, H.; Zhang, X.; Song, E.; Wang, Y.; Xu, T.; Li, Z. WFS1 Functions in ER Export of Vesicular Cargo Proteins in Pancreatic β-Cells. Nat. Commun. 2021, 12, 6996. [Google Scholar] [CrossRef]
- Tein, K.; Kasvandik, S.; Kõks, S.; Vasar, E.; Terasmaa, A. Prohormone Convertase 2 Activity Is Increased in the Hippocampus of Wfs1 Knockout Mice. Front. Mol. Neurosci. 2015, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Swift, R.G.; Polymeropoulos, M.H.; Torres, R.; Swift, M. Predisposition of Wolfram Syndrome Heterozygotes to Psychiatric Illness. Mol. Psychiatry 1998, 3, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Swift, R.G. Wolframin Mutations and Hospitalization for Psychiatric Illness. Mol. Psychiatry 2005, 10, 799–803. [Google Scholar] [CrossRef] [Green Version]
- Munshani, S.; Ibrahim, E.Y.; Domenicano, I.; Ehrlich, B.E. The Impact of Mutations in Wolframin on Psychiatric Disorders. Front. Pediatr. 2021, 9, 718132. [Google Scholar] [CrossRef]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diab. Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Haile, Y.; Carmine-Simmen, K.; Olechowski, C.; Kerr, B.; Bleackley, R.C.; Giuliani, F. Granzyme B-Inhibitor Serpina3n Induces Neuroprotection in Vitro and in Vivo. J. Neuroinflamm. 2015, 12, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Bai, Q.; Feng, J.; Zhao, B.; Duan, J.; Zhao, L.; Liu, N.; Ren, D.; Zou, S.; Chen, W. Neonatal Microglia and Proteinase Inhibitors-Treated Adult Microglia Improve Traumatic Brain Injury in Rats by Resolving the Neuroinflammation. Bioeng. Transl. Med. 2022, 7, e10249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Q.; Chen, D.; Zhao, W.; Wang, H.; Yang, M.; Xiang, Z.; Yuan, H. SerpinA3N Attenuates Ischemic Stroke Injury by Reducing Apoptosis and Neuroinflammation. CNS Neurosci. Ther. 2022, 28, 566–579. [Google Scholar] [CrossRef]
- Xi, Y.; Liu, M.; Xu, S.; Hong, H.; Chen, M.; Tian, L.; Xie, J.; Deng, P.; Zhou, C.; Zhang, L.; et al. Inhibition of SERPINA3N-Dependent Neuroinflammation Is Essential for Melatonin to Ameliorate Trimethyltin Chloride-Induced Neurotoxicity. J. Pineal Res. 2019, 67, e12596. [Google Scholar] [CrossRef]
- Murphy, C.E.; Kondo, Y.; Walker, A.K.; Rothmond, D.A.; Matsumoto, M.; Shannon Weickert, C. Regional, Cellular and Species Difference of Two Key Neuroinflammatory Genes Implicated in Schizophrenia. Brain. Behav. Immun. 2020, 88, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Chrast, R.; Verheijen, M.H.G.; Lemke, G. Complement Factors in Adult Peripheral Nerve: A Potential Role in Energy Metabolism. Neurochem. Int. 2004, 45, 353–359. [Google Scholar] [CrossRef]
- Ramaglia, V.; Daha, M.R.; Baas, F. The Complement System in the Peripheral Nerve: Friend or Foe? Mol. Immunol. 2008, 45, 3865–3877. [Google Scholar] [CrossRef]
- Spielman, L.; Winger, D.; Ho, L.; Aisen, P.S.; Shohami, E.; Pasinetti, G.M. Induction of the Complement Component C1qB in Brain of Transgenic Mice with Neuronal Overexpression of Human Cyclooxygenase-2. Acta Neuropathol. 2002, 103, 157–162. [Google Scholar] [CrossRef]
- Swanberg, M.; Duvefelt, K.; Diez, M.; Hillert, J.; Olsson, T.; Piehl, F.; Lidman, O. Genetically Determined Susceptibility to Neurodegeneration Is Associated with Expression of Inflammatory Genes. Neurobiol. Dis. 2006, 24, 67–88. [Google Scholar] [CrossRef]
- Kraft, A.D.; McPherson, C.A.; Harry, G.J. Association Between Microglia, Inflammatory Factors, and Complement with Loss of Hippocampal Mossy Fiber Synapses Induced by Trimethyltin. Neurotox. Res. 2016, 30, 53–66. [Google Scholar] [CrossRef]
- Wu, C.; Bendriem, R.M.; Freed, W.J.; Lee, C.-T. Transcriptome Analysis of Human Dorsal Striatum Implicates Attenuated Canonical WNT Signaling in Neuroinflammation and in Age-Related Impairment of Striatal Neurogenesis and Synaptic Plasticity. Restor. Neurol. Neurosci. 2021, 39, 247–266. [Google Scholar] [CrossRef]
- Ma, L.; Kulesskaya, N.; Võikar, V.; Tian, L. Differential Expression of Brain Immune Genes and Schizophrenia-Related Behavior in C57BL/6N and DBA/2J Female Mice. Psychiatry Res. 2015, 226, 211–216. [Google Scholar] [CrossRef]
- Careaga, M.; Schwartzer, J.; Ashwood, P. Inflammatory Profiles in the BTBR Mouse: How Relevant Are They to Autism Spectrum Disorders? Brain. Behav. Immun. 2015, 43, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Mutovina, A.; Ayriyants, K.; Mezhlumyan, E.; Ryabushkina, Y.; Litvinova, E.; Bondar, N.; Khantakova, J.; Reshetnikov, V. Unique Features of the Immune Response in BTBR Mice. Int. J. Mol. Sci. 2022, 23, 15577. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.-D.; Lai, L.B.; Zahurancik, W.J.; Gopalan, V. The Many Faces of RNA-Based RNase P, an RNA-World Relic. Trends Biochem. Sci. 2021, 46, 976–991. [Google Scholar] [CrossRef]
- Lipovich, L.; Dachet, F.; Cai, J.; Bagla, S.; Balan, K.; Jia, H.; Loeb, J.A. Activity-Dependent Human Brain Coding/Noncoding Gene Regulatory Networks. Genetics 2012, 192, 1133–1148. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Sun, Z.; Jia, H.; Luo, H.; Ye, X.; Wu, Q.; Xiong, Y.; Zhang, W.; Wan, J. Rpph1 Upregulates CDC42 Expression and Promotes Hippocampal Neuron Dendritic Spine Formation by Competing with MiR-330-5p. Front. Mol. Neurosci. 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, R.; Wang, L.; Tang, M.; Li, S.-R.; Liu, R.; Hu, X. LncRNA Rpph1 Protects Amyloid-β Induced Neuronal Injury in SK-N-SH Cells via MiR-122/Wnt1 Axis. Int. J. Neurosci. 2020, 130, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Liu, R.; Wang, L.; Tang, M.; Li, S.-R.; Hu, X. LncRNA RPPH1 Attenuates Aβ(25-35)-Induced Endoplasmic Reticulum Stress and Apoptosis in SH-SY5Y Cells via MiR-326/PKM2. Int. J. Neurosci. 2021, 131, 425–432. [Google Scholar] [CrossRef]
- Huang, H.-S.; Cheung, I.; Akbarian, S. RPP25 Is Developmentally Regulated in Prefrontal Cortex and Expressed at Decreased Levels in Autism Spectrum Disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2010, 3, 153–161. [Google Scholar] [CrossRef]
- Song, M.-G.; Bail, S.; Kiledjian, M. Multiple Nudix Family Proteins Possess MRNA Decapping Activity. RNA 2013, 19, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Grudzien-Nogalska, E.; Hamilton, K.; Jiao, X.; Yang, J.; Tong, L.; Kiledjian, M. Mammalian Nudix Proteins Cleave Nucleotide Metabolite Caps on RNAs. Nucleic Acids Res. 2020, 48, 6788–6798. [Google Scholar] [CrossRef]
- Shumar, S.A.; Kerr, E.W.; Geldenhuys, W.J.; Montgomery, G.E.; Fagone, P.; Thirawatananond, P.; Saavedra, H.; Gabelli, S.B.; Leonardi, R. Nudt19 Is a Renal CoA Diphosphohydrolase with Biochemical and Regulatory Properties That Are Distinct from the Hepatic Nudt7 Isoform. J. Biol. Chem. 2018, 293, 4134–4148. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.W.; Shumar, S.A.; Leonardi, R. Nudt8 Is a Novel CoA Diphosphohydrolase That Resides in the Mitochondria. FEBS Lett. 2019, 593, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Görigk, S.; Ouwens, D.M.; Kuhn, T.; Altenhofen, D.; Binsch, C.; Damen, M.; Khuong, J.M.-A.; Kaiser, K.; Knebel, B.; Vogel, H.; et al. Nudix Hydrolase NUDT19 Regulates Mitochondrial Function and ATP Production in Murine Hepatocytes. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2022, 1867, 159153. [Google Scholar] [CrossRef]
- Lan, C.; Wang, Y.; Su, X.; Lu, J.; Ma, S. LncRNA LINC00958 Activates MTORC1/P70S6K Signalling Pathway to Promote Epithelial-Mesenchymal Transition Process in the Hepatocellular Carcinoma. Cancer Investig. 2021, 39, 539–549. [Google Scholar] [CrossRef]
- Arisi, I.; D’Onofrio, M.; Brandi, R.; Felsani, A.; Capsoni, S.; Drovandi, G.; Felici, G.; Weitschek, E.; Bertolazzi, P.; Cattaneo, A. Gene Expression Biomarkers in the Brain of a Mouse Model for Alzheimer’s Disease: Mining of Microarray Data by Logic Classification and Feature Selection. J. Alzheimer’s Dis. 2011, 24, 721–738. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Yang, C.; Chen, J.; Gong, W.; Yi, F.; Liao, W.; Huang, R.; Xie, L.; Zhou, J. Proteomic Insights into Susceptibility and Resistance to Chronic-Stress-Induced Depression or Anxiety in the Rat Striatum. Front. Mol. Biosci. 2021, 8, 730473. [Google Scholar] [CrossRef]
- Polovitskaya, M.M.; Barbini, C.; Martinelli, D.; Harms, F.L.; Cole, F.S.; Calligari, P.; Bocchinfuso, G.; Stella, L.; Ciolfi, A.; Niceta, M.; et al. A Recurrent Gain-of-Function Mutation in CLCN6, Encoding the ClC-6 Cl−/H+-Exchanger, Causes Early-Onset Neurodegeneration. Am. J. Hum. Genet. 2020, 107, 1062–1077. [Google Scholar] [CrossRef]
- Poët, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.C.; Planells-Cases, R.; et al. Lysosomal Storage Disease upon Disruption of the Neuronal Chloride Transport Protein ClC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859. [Google Scholar] [CrossRef]
- Pressey, S.N.R.; O’Donnell, K.J.; Stauber, T.; Fuhrmann, J.C.; Tyynelä, J.; Jentsch, T.J.; Cooper, J.D. Distinct Neuropathologic Phenotypes after Disrupting the Chloride Transport Proteins ClC-6 or ClC-7/Ostm1. J. Neuropathol. Exp. Neurol. 2010, 69, 1228–1246. [Google Scholar] [CrossRef] [Green Version]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an Inner Mitochondrial Membrane Protein, Is Essential for Maintaining Crista Integrity and Mitochondrial Function. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef] [Green Version]
- Lionel, A.C.; Crosbie, J.; Barbosa, N.; Goodale, T.; Thiruvahindrapuram, B.; Rickaby, J.; Gazzellone, M.; Carson, A.R.; Howe, J.L.; Wang, Z.; et al. Rare Copy Number Variation Discovery and Cross-Disorder Comparisons Identify Risk Genes for ADHD. Sci. Transl. Med. 2011, 3, 95ra75. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.J.; Ka, M.; Jung, E.-M.; Smith, A.L.; Kim, W.-Y. The Role of MACF1 in Nervous System Development and Maintenance. Semin. Cell Dev. Biol. 2017, 69, 9–17. [Google Scholar] [CrossRef]
- Ka, M.; Jung, E.-M.; Mueller, U.; Kim, W.-Y. MACF1 Regulates the Migration of Pyramidal Neurons via Microtubule Dynamics and GSK-3 Signaling. Dev. Biol. 2014, 395, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Dobyns, W.B.; Aldinger, K.A.; Ishak, G.E.; Mirzaa, G.M.; Timms, A.E.; Grout, M.E.; Dremmen, M.H.G.; Schot, R.; Vandervore, L.; van Slegtenhorst, M.A.; et al. MACF1 Mutations Encoding Highly Conserved Zinc-Binding Residues of the GAR Domain Cause Defects in Neuronal Migration and Axon Guidance. Am. J. Hum. Genet. 2018, 103, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Courchesne, E.; Gazestani, V.H.; Lewis, N.E. Prenatal Origins of ASD: The When, What, and How of ASD Development. Trends Neurosci. 2020, 43, 326–342. [Google Scholar] [CrossRef]
- Kanamarlapudi, V. Exchange Factor EFA6R Requires C-Terminal Targeting to the Plasma Membrane to Promote Cytoskeletal Rearrangement through the Activation of ADP-Ribosylation Factor 6 (ARF6). J. Biol. Chem. 2014, 289, 33378–33390. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.A.; Jia, T.; Pinzón, J.H.; Acevedo, S.F.; Ojelade, S.A.; Xu, B.; Tay, N.; Desrivières, S.; Hernandez, J.L.; Banaschewski, T.; et al. The Arf6 Activator Efa6/PSD3 Confers Regional Specificity and Modulates Ethanol Consumption in Drosophila and Humans. Mol. Psychiatry 2018, 23, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Liang, H.; Chen, Y.; Qin, Q.; Wei, Y.; Liang, Z. Related Network and Differential Expression Analyses Identify Nuclear Genes and Pathways in the Hippocampus of Alzheimer Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919311. [Google Scholar] [CrossRef]
- Dong, P.; Xu, Q.; An, Y.; Zhou, B.-R.; Lu, P.; Liu, R.-C.; Xu, X. A Novel 1.0 Mb Duplication of Chromosome 8p22-21.3 in a Patient with Autism Spectrum Disorder. Child Neurol. Open 2015, 2, 1–6. [Google Scholar] [CrossRef]
- Gundelfinger, E.D.; Karpova, A.; Pielot, R.; Garner, C.C.; Kreutz, M.R. Organization of Presynaptic Autophagy-Related Processes. Front. Synaptic Neurosci. 2022, 14, 829354. [Google Scholar] [CrossRef]
- Hoffmann-Conaway, S.; Brockmann, M.M.; Schneider, K.; Annamneedi, A.; Rahman, K.A.; Bruns, C.; Textoris-Taube, K.; Trimbuch, T.; Smalla, K.-H.; Rosenmund, C.; et al. Parkin Contributes to Synaptic Vesicle Autophagy in Bassoon-Deficient Mice. Elife 2020, 9, e56590. [Google Scholar] [CrossRef]
- Montenegro-Venegas, C.; Guhathakurta, D.; Pina-Fernandez, E.; Andres-Alonso, M.; Plattner, F.; Gundelfinger, E.D.; Fejtova, A. Bassoon Controls Synaptic Vesicle Release via Regulation of Presynaptic Phosphorylation and CAMP. EMBO Rep. 2022, 23, e53659. [Google Scholar] [CrossRef]
- Annamneedi, A.; Caliskan, G.; Müller, S.; Montag, D.; Budinger, E.; Angenstein, F.; Fejtova, A.; Tischmeyer, W.; Gundelfinger, E.D.; Stork, O. Ablation of the Presynaptic Organizer Bassoon in Excitatory Neurons Retards Dentate Gyrus Maturation and Enhances Learning Performance. Brain Struct. Funct. 2018, 223, 3423–3445. [Google Scholar] [CrossRef] [Green Version]
- Annamneedi, A.; Del Angel, M.; Gundelfinger, E.D.; Stork, O.; Çalışkan, G. The Presynaptic Scaffold Protein Bassoon in Forebrain Excitatory Neurons Mediates Hippocampal Circuit Maturation: Potential Involvement of TrkB Signalling. Int. J. Mol. Sci. 2021, 22, 7944. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon Proteinopathy Drives Neurodegeneration in Multiple Sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef]
- Chen, C.-H.; Huang, Y.-S.; Liao, D.-L.; Huang, C.-Y.; Lin, C.-H.; Fang, T.-H. Identification of Rare Mutations of Two Presynaptic Cytomatrix Genes BSN and PCLO in Schizophrenia and Bipolar Disorder. J. Pers. Med. 2021, 11, 1057. [Google Scholar] [CrossRef]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram Syndrome 1) Gene Product: Predominant Subcellular Localization to Endoplasmic Reticulum in Cultured Cells and Neuronal Expression in Rat Brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Marshall, B.A.; Permutt, M.A.; Paciorkowski, A.R.; Hoekel, J.; Karzon, R.; Wasson, J.; Viehover, A.; White, N.H.; Shimony, J.S.; Manwaring, L.; et al. Phenotypic Characteristics of Early Wolfram Syndrome. Orphanet J. Rare Dis. 2013, 8, 64. [Google Scholar] [CrossRef]
- Bischoff, A.N.; Reiersen, A.M.; Buttlaire, A.; Al-Lozi, A.; Doty, T.; Marshall, B.A.; Hershey, T. Selective Cognitive and Psychiatric Manifestations in Wolfram Syndrome. Orphanet J. Rare Dis. 2015, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Moy, S.S.; Nadler, J.J.; Poe, M.D.; Nonneman, R.J.; Young, N.B.; Koller, B.H.; Crawley, J.N.; Duncan, G.E.; Bodfish, J.W. Development of a Mouse Test for Repetitive, Restricted Behaviors: Relevance to Autism. Behav. Brain Res. 2008, 188, 178–194. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Abrams, D.N.; Zhang, J.Y.; Weber, M.D.; Katz, A.M.; Clarke, A.M.; Silverman, J.L.; Crawley, J.N. Low Sociability in BTBR T+tf/J Mice Is Independent of Partner Strain. Physiol. Behav. 2012, 107, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, H. Somatosensorimotor and Odor Modification, Along with Serotonergic Processes Underlying the Social Deficits in BTBR T+ Itpr3(Tf)/J and BALB/CJ Mouse Models of Autism. Neuroscience 2020, 445, 144–162. [Google Scholar] [CrossRef]
- Frye, C.A.; Llaneza, D.C. Corticosteroid and Neurosteroid Dysregulation in an Animal Model of Autism, BTBR Mice. Physiol. Behav. 2010, 100, 264–267. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.L.; Yang, M.; Turner, S.M.; Katz, A.M.; Bell, D.B.; Koenig, J.I.; Crawley, J.N. Low Stress Reactivity and Neuroendocrine Factors in the BTBR T+tf/J Mouse Model of Autism. Neuroscience 2010, 171, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Benno, R.; Smirnova, Y.; Vera, S.; Liggett, A.; Schanz, N. Exaggerated Responses to Stress in the BTBR T+tf/J Mouse: An Unusual Behavioral Phenotype. Behav. Brain Res. 2009, 197, 462–465. [Google Scholar] [CrossRef]
- Gould, G.G.; Burke, T.F.; Osorio, M.D.; Smolik, C.M.; Zhang, W.Q.; Onaivi, E.S.; Gu, T.-T.; DeSilva, M.N.; Hensler, J.G. Enhanced Novelty-Induced Corticosterone Spike and Upregulated Serotonin 5-HT1A and Cannabinoid CB1 Receptors in Adolescent BTBR Mice. Psychoneuroendocrinology 2014, 39, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.; Khanbabaei, M.; Murari, K.; Rho, J.M. Disruption of Visual Circuit Formation and Refinement in a Mouse Model of Autism. Autism Res. Off. J. Int. Soc. Autism Res. 2017, 10, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Ou, J.-N.; Cao, L.-F.; Peng, X.-Q.; Li, Y.-M.; Tian, Y.-Q. The Autism-Related LncRNA MSNP1AS Regulates Moesin Protein to Influence the RhoA, Rac1, and PI3K/Akt Pathways and Regulate the Structure and Survival of Neurons. Autism Res. Off. J. Int. Soc. Autism Res. 2020, 13, 2073–2082. [Google Scholar] [CrossRef]
- Heo, Y.; Zhang, Y.; Gao, D.; Miller, V.M.; Lawrence, D.A. Aberrant Immune Responses in a Mouse with Behavioral Disorders. PLoS ONE 2011, 6, e20912. [Google Scholar] [CrossRef] [Green Version]
- Bakheet, S.A.; Alzahrani, M.Z.; Nadeem, A.; Ansari, M.A.; Zoheir, K.M.A.; Attia, S.M.; Al-Ayadhi, L.Y.; Ahmad, S.F. Resveratrol Treatment Attenuates Chemokine Receptor Expression in the BTBR T+tf/J Mouse Model of Autism. Mol. Cell. Neurosci. 2016, 77, 1–10. [Google Scholar] [CrossRef]
- Uddin, M.N.; Yao, Y.; Mondal, T.; Matala, R.; Manley, K.; Lin, Q.; Lawrence, D.A. Immunity and Autoantibodies of a Mouse Strain with Autistic-like Behavior. Brain Behav. Immun. - Health 2020, 4, 100069. [Google Scholar] [CrossRef]
- Palmer-Aronsten, B.; Sheedy, D.; McCrossin, T.; Kril, J. An International Survey of Brain Banking Operation and Characterization Practices. Biopreserv. Biobank. 2016, 14, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of Stress throughout the Lifespan on the Brain, Behaviour and Cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Age | Brain Region | Method | Threshold | Raw Data | Reference |
|---|---|---|---|---|---|---|
| BTBR T + Itpr3tf/J (BTBR)/C57BL/6J | 12 w | Hippocampus | RNA-seq | p value < 0.05 * | PRJNA533538 | [38] |
| BTBR T+ Itpr3tf/J (BTBR)/C57BL/6J | 3–5 m | Hippocampus | Microarray | Rank Product (RP) non-parametric method was used | GSE81501 | [39] |
| BTBR T + Itprtf/J mice/C57BL/6J | 4 m | Hippocampus and cortex | Microarray | Z-ratio value of ±1.50 and/or a Z-test value p < 0.05 | N/A | [40] |
| BTBR/B6 | 8 w | Cortex | RNA-seq | |log2FC| ≥ 1 and padj ≤ 0.05 | N/A | [41] |
| BTBR T + tf/J/C57BL/6J | 8–10 w | Striatum (bregma −0.58–1.53). | RNA-seq | p value < 0.05 * | GSE138539 | [42] |
| BTBRTF/ArtRbrc mice Compared to C57BL/6J Mice | 7 w | Striatum + Cortex | Microarray | padj ≤ 0.05 | GSE156646 | [34] |
| BTBR T + Itprtf/J mice/C57BL/6J | 4 m | Hippocampus | iTRAQ LC–MS/MS | fold change > 0.2 | N/A | [40] |
| BTBR T + Itprtf/J mice/C57BL/6J | 8 w | Cortex | iTRAQ LC–MS/MS | fold change > 0.5, p < 0.05 | N/A | [43] |
| Disease | Type of Review | Data ID | Method | Threshold | Reference |
|---|---|---|---|---|---|
| ASD | Meta-analysis | GSE28475 | Microarray/RNA-seq | p < 0.05 | [51] |
| GSE28521 | |||||
| GSE36192 | |||||
| ASD | Meta-analysis | GSE64018 | RNA-seq | p < 0.05 | [52] |
| GSE30573 | |||||
| SCZ | Meta-analysis | GSE17612 | Microarray | p < 0.05 | [53] |
| GSE21935 | |||||
| GSE25673 | |||||
| GSE12649 | |||||
| GSE21338 | |||||
| SCZ | Meta-analysis | GSE17612 | Microarray | q-value < 0.05 | [54] |
| GSE21138 | |||||
| +and four others |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisaretova, P.; Tsybko, A.; Bondar, N.; Reshetnikov, V. Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies. Biomedicines 2023, 11, 289. https://doi.org/10.3390/biomedicines11020289
Kisaretova P, Tsybko A, Bondar N, Reshetnikov V. Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies. Biomedicines. 2023; 11(2):289. https://doi.org/10.3390/biomedicines11020289
Chicago/Turabian StyleKisaretova, Polina, Anton Tsybko, Natalia Bondar, and Vasiliy Reshetnikov. 2023. "Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies" Biomedicines 11, no. 2: 289. https://doi.org/10.3390/biomedicines11020289
APA StyleKisaretova, P., Tsybko, A., Bondar, N., & Reshetnikov, V. (2023). Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies. Biomedicines, 11(2), 289. https://doi.org/10.3390/biomedicines11020289