

An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease
 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
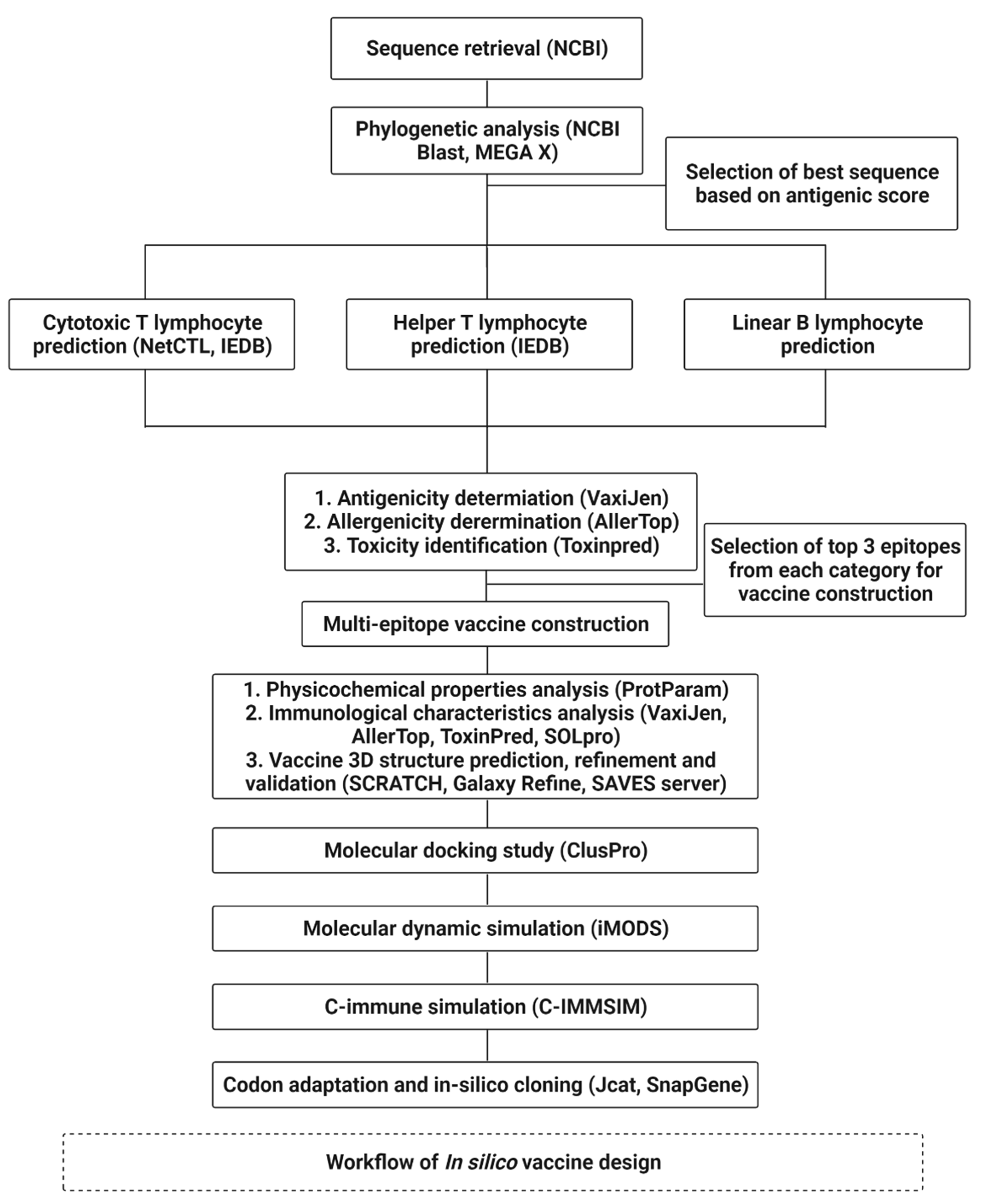
2. Methodology
2.1. Sequence Data Retrieval and Phylogenetic Analysis
2.2. B- and T-Cell Lymphocyte Prediction
2.3. Development of Vaccine Based on Cytotoxicity, Allergenicity, and Antigenicity
2.4. Prediction and Molecular Docking of Vaccines
2.5. Simulation of the Docked Complex
2.6. C-Immune Simulation
2.7. Codon Adaptation and In Silico Cloning
3. Results
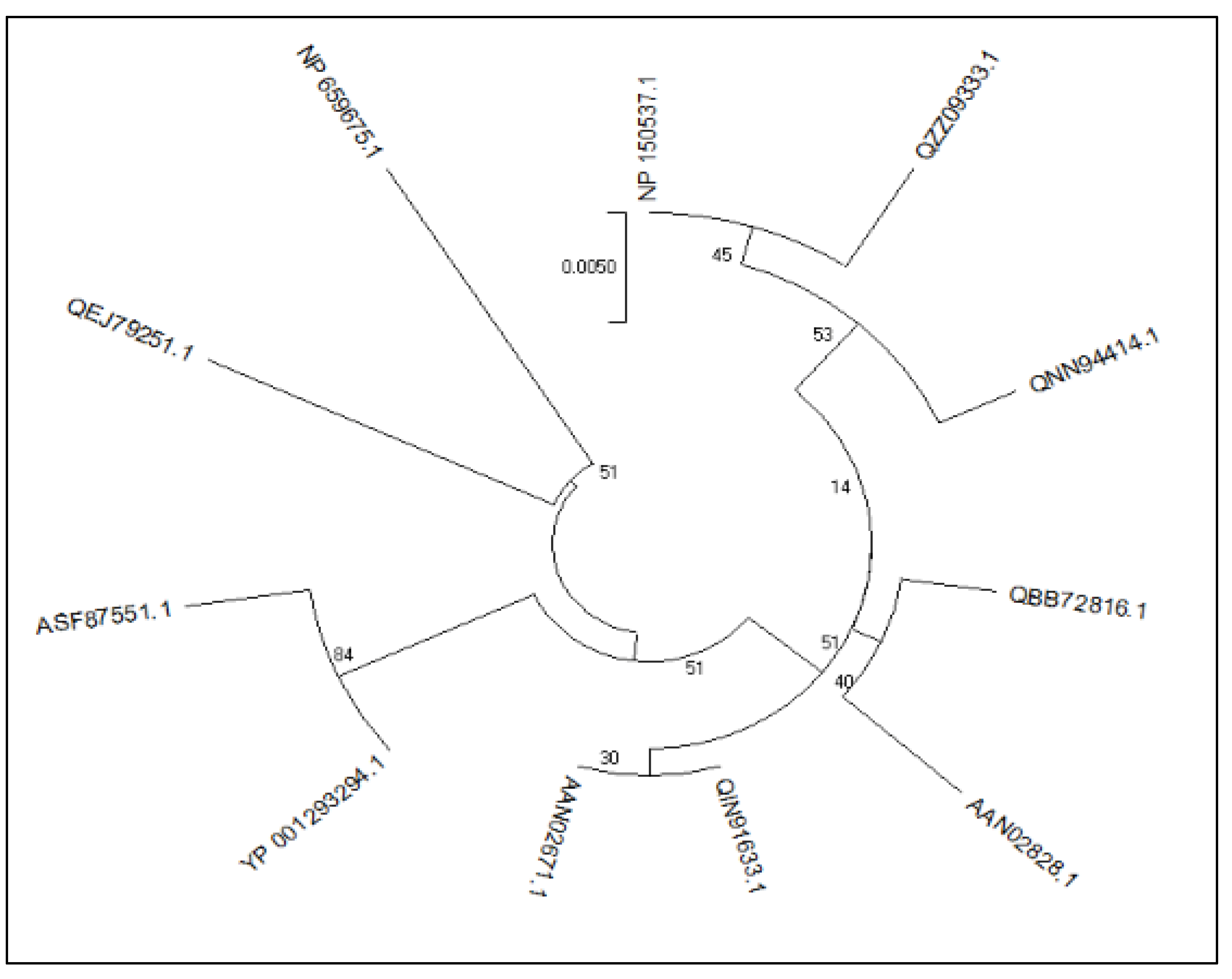
3.1. Phylogenetic Analysis of the Retrieved Sequences
3.2. Physiochemical Properties
3.3. Secondary Structure of Lumpy Skin Protein
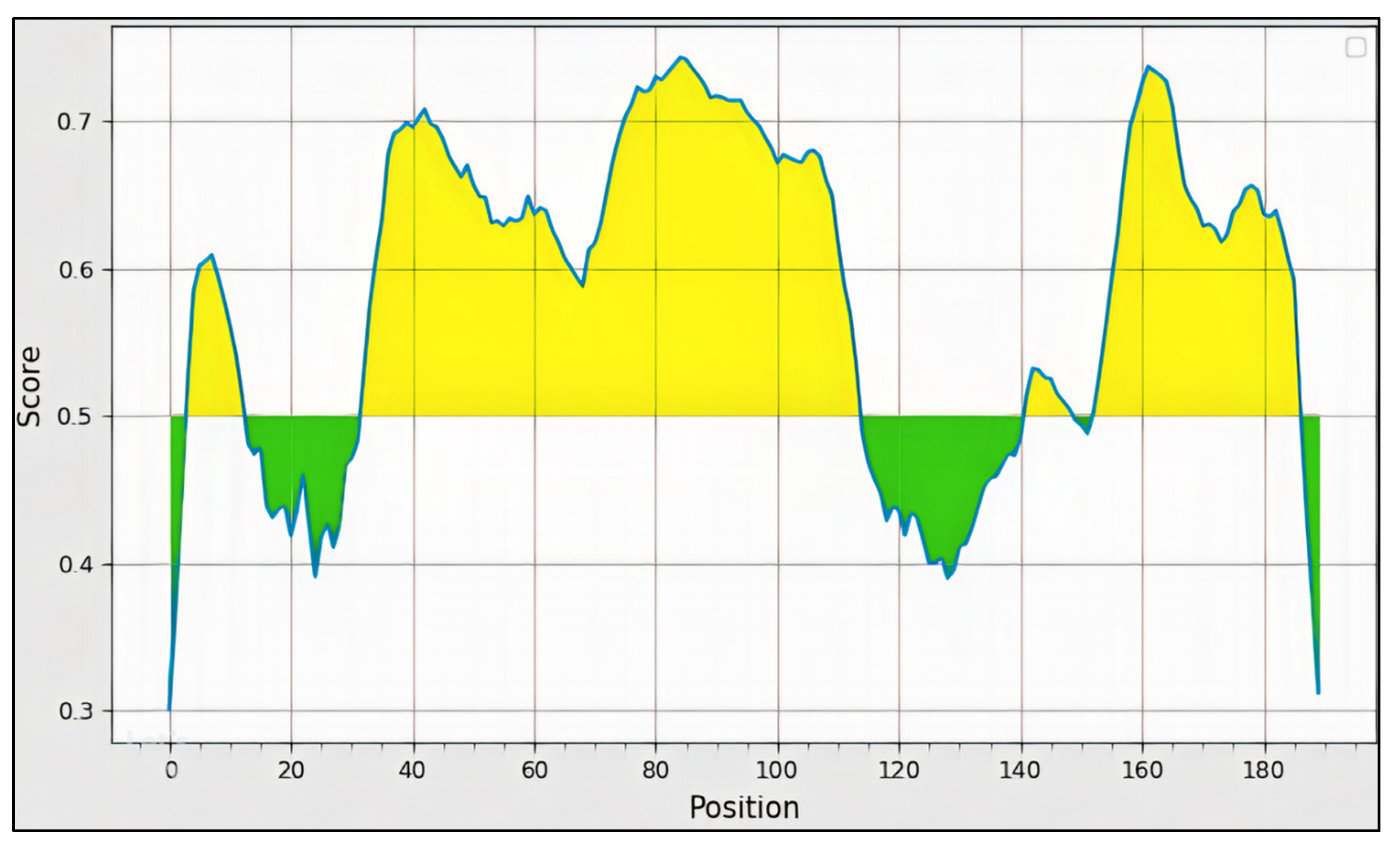
3.4. B-Cell Epitope Prediction
3.5. T-Cell Epitope Prediction
3.5.1. MHC-1 Epitopes
3.5.2. MHC-II Epitopes
3.6. Population Coverage
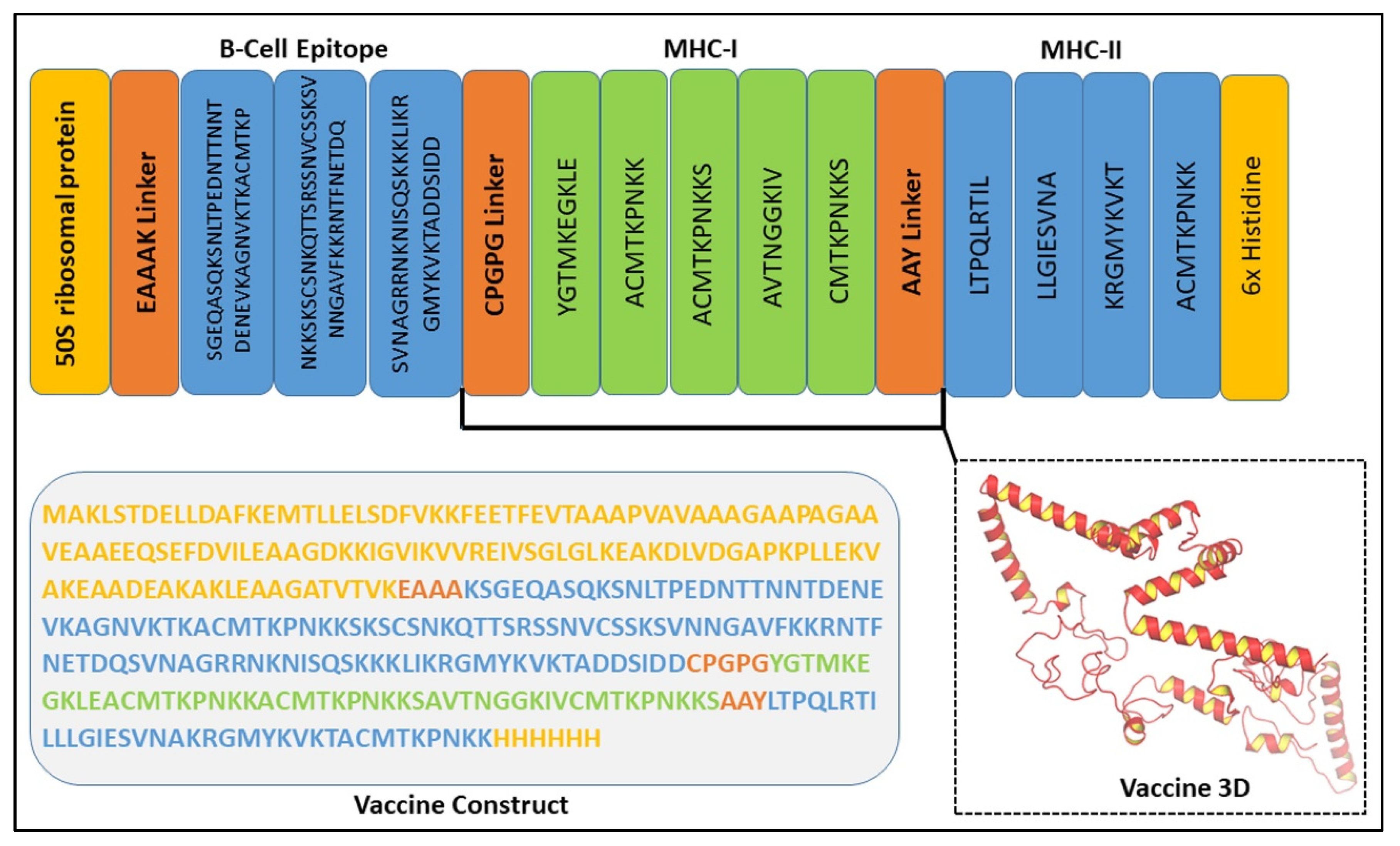
3.7. Assessment of the Chimeric Vaccine Construct
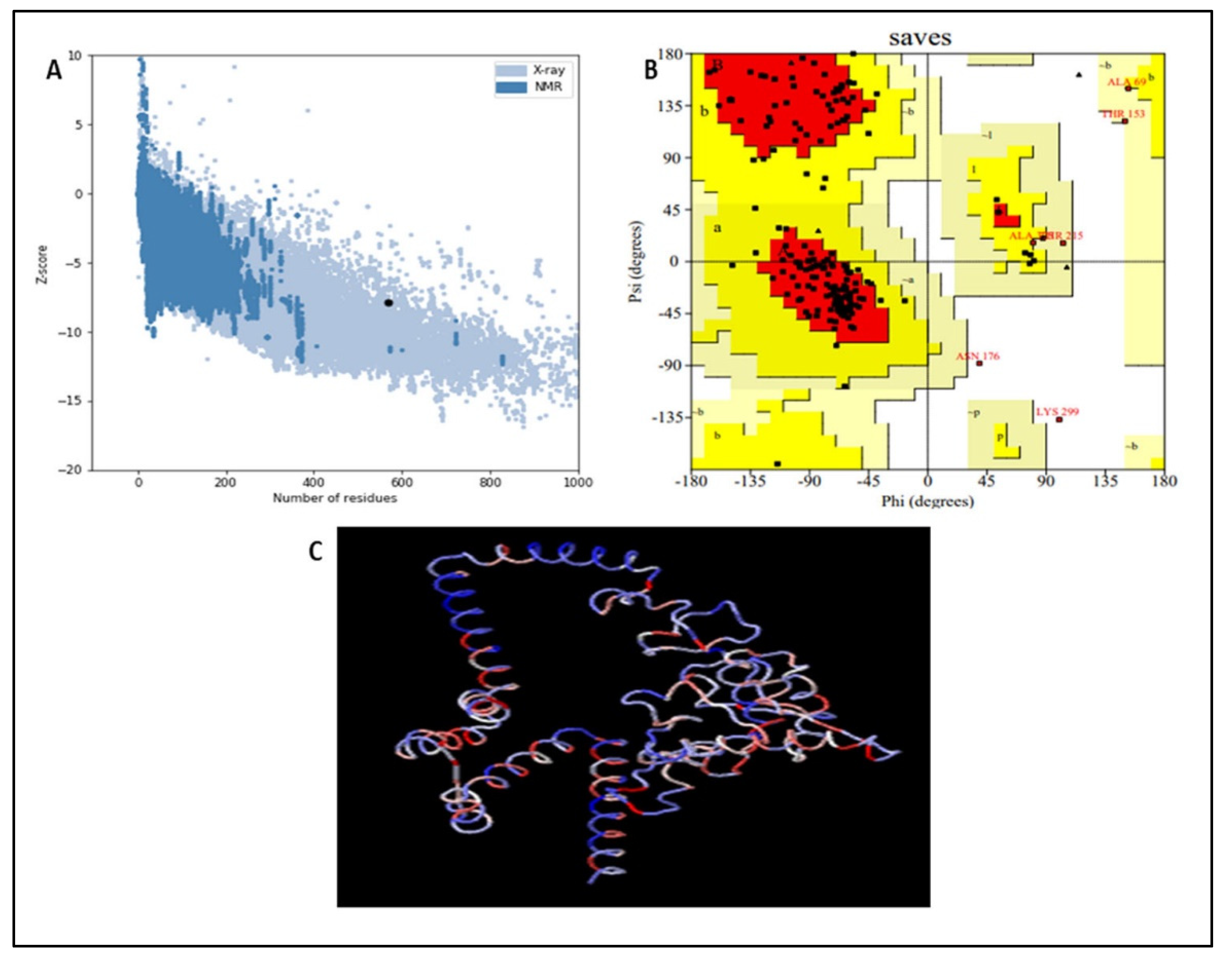
3.8. Vaccine Refinement and Validation
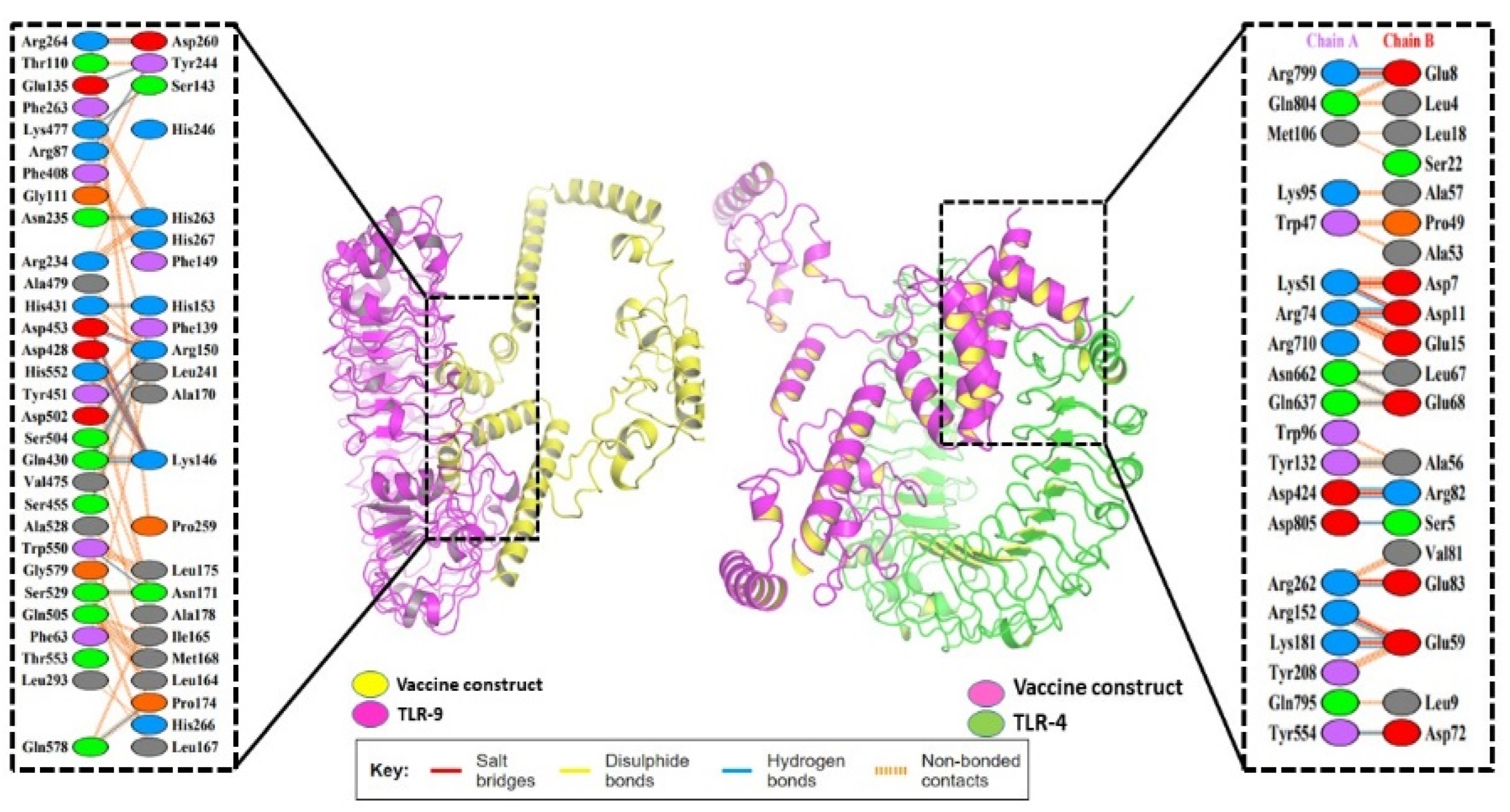
3.9. Molecular Docking of the Vaccine
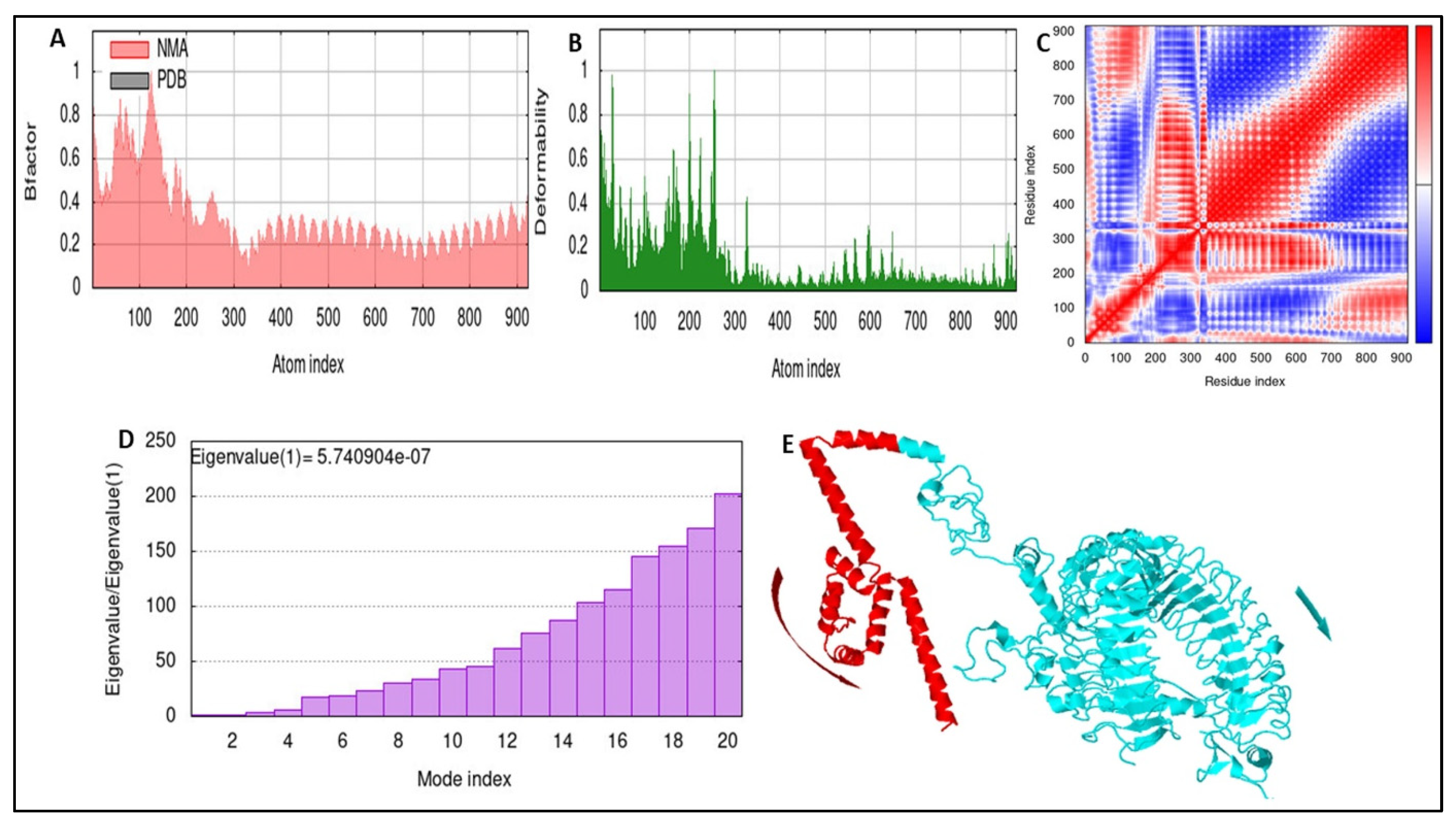
3.10. Stability of the Docked Complex
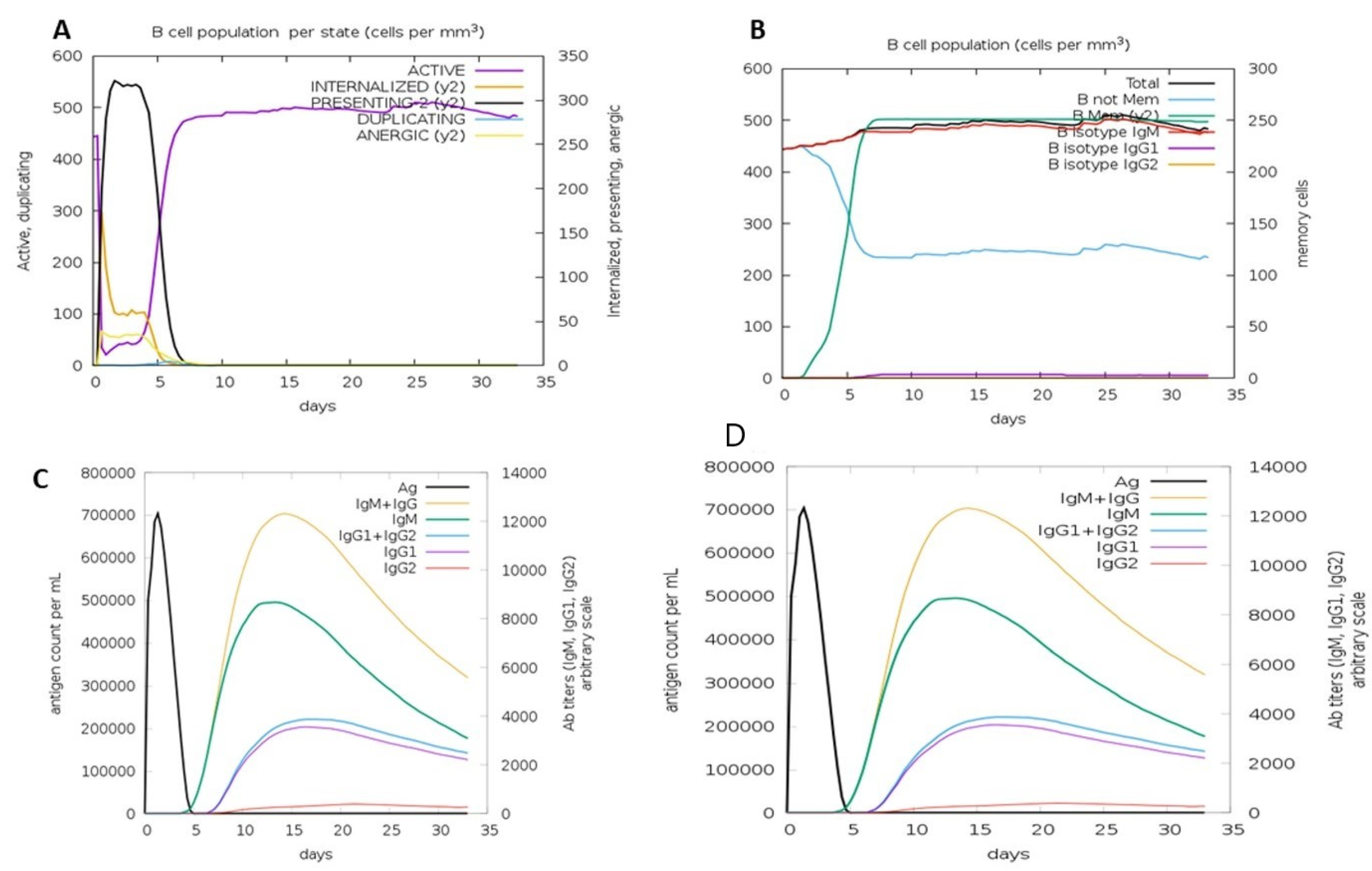
3.11. Immune Simulation
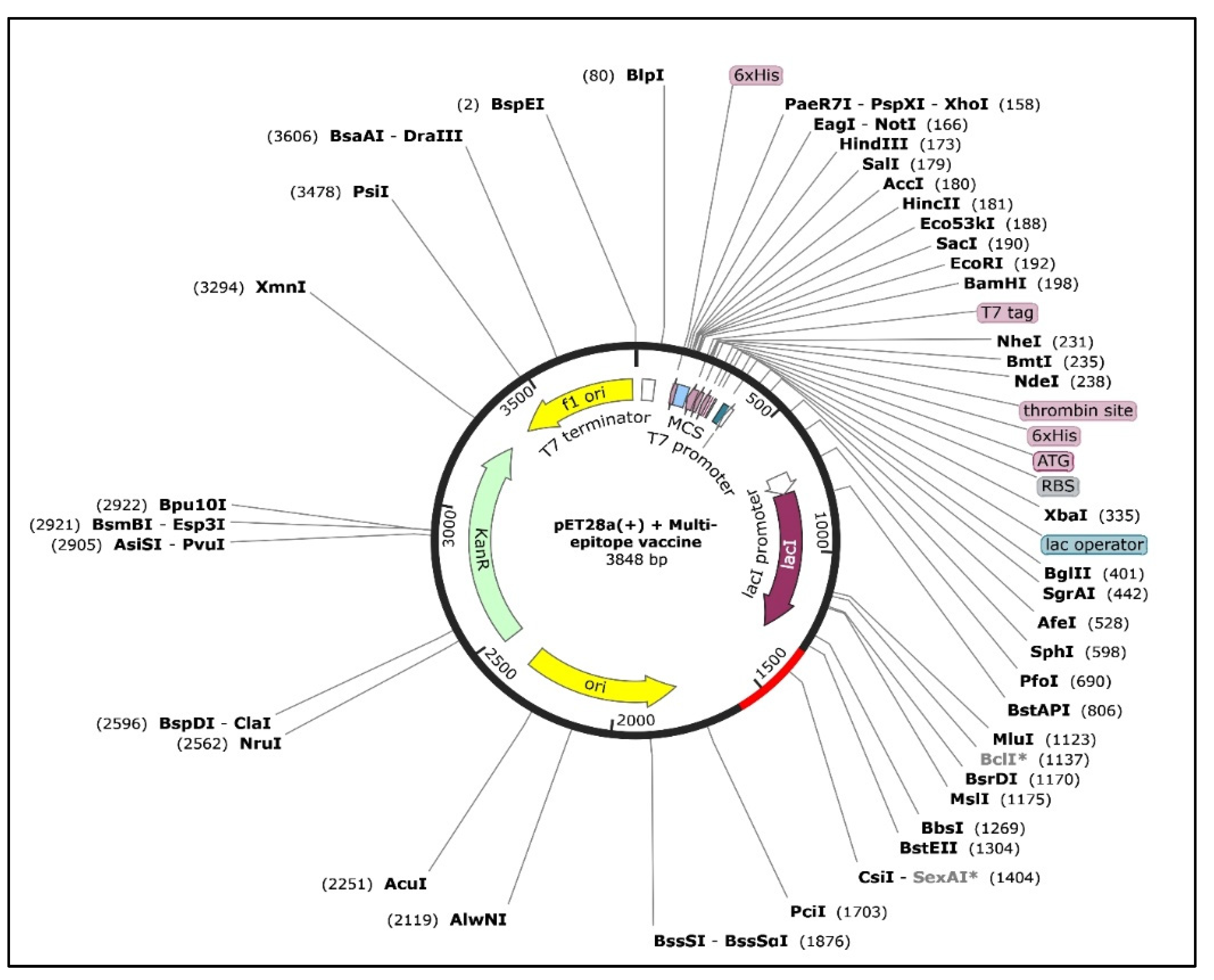
3.12. In Silico Cloning
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jongejan, F.; Uilenberg, G. The global importance of ticks. Parasitology 2004, 129, S3–S14. [Google Scholar] [CrossRef]
- Lubinga, J.C.; Tuppurainen, E.S.M.; Coetzer, J.A.W.; Stoltsz, W.H.; Venter, E.H. Evidence of lumpy skin disease virus over-wintering by transstadial persistence in Amblyomma hebraeum and transovarial persistence in Rhipicephalus decoloratus ticks. Exp. Appl. Acarol. 2014, 62, 77–90. [Google Scholar] [CrossRef]
- Swiswa, S.; Masocha, M.; Pfukenyi, D.M.; Dhliwayo, S.; Chikerema, S.M. Long-term changes in the spatial distribution of lumpy skin disease hotspots in Zimbabwe. Trop. Anim. Health Prod. 2016, 49, 195–199. [Google Scholar] [CrossRef]
- Tuppurainen, E.S.M.; Stoltsz, W.H.; Troskie, M.; Wallace, D.B.; Oura, C.A.L.; Mellor, P.S.; Coetzer, J.A.W.; Venter, E. A Potential Role for Ixodid (Hard) Tick Vectors in the Transmission of Lumpy Skin Disease Virus in Cattle. Transbound. Emerg. Dis. 2011, 58, 93–104. [Google Scholar] [CrossRef]
- Angelova, T.; Yordanova, D.; Krastanov, J.; Miteva, D.; Kalaydhziev, G.; Karabashev, V.; Mihaylova, M.; Marutsov, P.; Ivanov, N. Quantitative and qualitative changes in milk yield and cheese-making properties of milk in cows vac-cinated against lumpy skin disease. Maced. J. Anim. Sci. 2018, 8, 89–95. [Google Scholar] [CrossRef]
- Yousefi, P.S.; Mardani, K.; Dalir-Naghadeh, B.; Jalilzadeh-Amin, G. Epidemiological Study of Lumpy Skin Disease Outbreaks in North-western Iran. Transbound. Emerg. Dis. 2016, 64, 1782–1789. [Google Scholar] [CrossRef]
- Gelaye, E.; Lamien, C.E. Sheep and goat pox. In Transboundary Animal Diseases in Sahelian Africa and Connected Regions; Springer: Berlin/Heidelberg, Germany, 2019; pp. 289–303. [Google Scholar]
- European Food Safety Authority (EFSA); Calistri, P.; DeClercq, K.; Gubbins, S.; Klement, E.; Stegeman, A.; Abrahantes, J.C.; Antoniou, S.; Broglia, A.; Gogin, A. Lumpy skin disease: III. Data collection and analysis. EFSA J. 2019, 17, e05638. [Google Scholar]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Hemadri, D.; Sood, R.; Bal, G.C.; Nayak, M.K.; Pradhan, S.K.; Singh, V.P. Lumpy skin disease (LSD) outbreaks in cattle in Odisha state, India in August 2019: Epidemiological features and molecular studies. Transbound. Emerg. Dis. 2020, 67, 2408–2422. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA); Calistri, P.; De Clercq, K.; Gubbins, S.; Klement, E.; Stegeman, A.; Abrahantes, J.C.; Marojevic, D.; Antoniou, S.-E.; Broglia, A. Lumpy skin disease epidemiological report IV: Data collection and analysis. EFSA J. 2020, 18, e06010. [Google Scholar]
- Bhanuprakash, V.; Indrani, B.K.; Hosamani, M.; Singh, R.K. The current status of sheep pox disease. Comp. Immunol. Microbiol. Infect. Dis. 2006, 29, 27–60. [Google Scholar] [CrossRef]
- Abdulqa, H.Y.; Rahman, H.S.; Dyary, H.O.; Othman, H.H. Lumpy skin disease. Reprod. Immunol. Open Access 2016, 1, 2476-1974. [Google Scholar] [CrossRef]
- Shahab, M.; Hayat, C.; Sikandar, R.; Zheng, G.; Akter, S. In silico designing of a multi-epitope vaccine against Burkholderia pseudomallei: Reverse vaccinology and immunoinformatics. J. Genet. Eng. Biotechnol. 2022, 20, 100. [Google Scholar] [CrossRef]
- Akter, S.; Shahab, M.; Sarkar, M.H.; Hayat, C.; Banu, T.A.; Goswami, B.; Jahan, I.; Osman, E.; Uzzaman, M.S.; Habib, A.; et al. Immunoinformatics approach to epitope-based vaccine design against the SARS-CoV-2 in Bangladeshi patients. J. Genet. Eng. Biotechnol. 2022, 20, 136. [Google Scholar] [CrossRef]
- Yadav, A.; Ojha, M.D.; Hariprasad, P. Computational studies evidenced the potential of steroidal lactone to disrupt surface interaction of SARS-CoV-2 spike protein and hACE2. Comput. Biol. Med. 2022, 146, 105598. [Google Scholar] [CrossRef]
- Le Mercier, P.; Jacob, Y.; Tanner, K.; Tordo, N. A Novel Expression Cassette of Lyssavirus Shows that the Distantly Related Mokola Virus Can Rescue a Defective Rabies Virus Genome. J. Virol. 2002, 76, 2024–2027. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ismail, S.; Ahmad, S.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Hayat, C.; Shahab, M.; Khan, S.A.; Liang, C.; Duan, X.; Khan, H.; Zheng, G.; Ul-Haq, Z. Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat monkeypox. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Quiros-Fernandez, I.; Poorebrahim, M.; Fakhr, E.; Cid-Arregui, A. Immunogenic T cell epitopes of SARS-CoV-2 are recognized by circulating memory and naïve CD8 T cells of unexposed individuals. EBioMedicine 2021, 72, 103610. [Google Scholar] [CrossRef]
- Flower, D.R.; Doytchinova, I.; Zaharieva, N.; Dimitrov, I. Immunogenicity Prediction by VaxiJen: A Ten Year Overview. J. Proteom. Bioinform. 2017, 10, 298–310. [Google Scholar] [CrossRef]
- Hizbullah; Nazir, Z.; Afridi, S.G.; Shah, M.; Shams, S.; Khan, A. Reverse vaccinology and subtractive genomics-based putative vaccine targets identification for Burkholderia pseudomallei Bp1651. Microb. Pathog. 2018, 125, 219–229. [Google Scholar] [CrossRef]
- Muneeba, A. Effect of spiritual intelligence on effective change management: A review of selected researches. Electron. Res. J. Soc. Sci. Humanit. 2019, 1, 30–47. [Google Scholar]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy that Enhances CD4+ T Help in BALB/c Mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef]
- Almofti, Y.A.; Abd-elrahman, K.A.; Eltili, E.E.M. Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Immunol. 2021, 22, 1–20. [Google Scholar] [CrossRef]
- Saha, R.; Prasad, B.V. In silico approach for designing of a multi-epitope based vaccine against novel Coronavirus (SARS-COV-2). BioRxiv 2020. [Google Scholar] [CrossRef]
- Abdi, S.A.H.; Ali, A.; Sayed, S.F.; Abutahir; Ali, A.; Alam, P. Multi-Epitope-Based Vaccine Candidate for Monkeypox: An in Silico Approach. Vaccines 2022, 10, 1564. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Sami, S.A.; Marma, K.K.S.; Mahmud, S.; Khan, A.N.; Albogami, S.; El-Shehawi, A.M.; Rakib, A.; Chakraborty, A.; Mohiuddin, M.; Dhama, K.; et al. Designing of a multi-epitope vaccine against the structural proteins of marburg virus exploiting the im-munoinformatics approach. ACS Omega 2021, 6, 32043–32071. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.-B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Arumugam, S.; Varamballi, P. In-silico design of envelope based multi-epitope vaccine candidate against Kyasanur forest disease virus. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.S.; Huber, S.; Alcantara-Neves, N.M.; Asam, C.; Silveira, E.F.; de Andrade Belitardo, E.M.M.; Aglas, L.; Wallner, M.; Gadermaier, G.; Briza, P. N-terminal peptide deletion influences immunological and structural features of Blo t 5. Allergy 2020, 75, 1503–1507. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.; Flower, D.R. Bioinformatic Approach for Identifying Parasite and Fungal Candidate Subunit Vaccines. Open Vaccine J. 2008, 1, 22–26. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed]
- Didelon, M.; Khafif, M.; Godiard, L.; Barbacci, A.; Raffaele, S. Patterns of Sequence and Expression Diversification Associate Members of the PADRE Gene Family with Response to Fungal Pathogens. Front. Genet. 2020, 11, 491. [Google Scholar] [CrossRef]
- Aslam, M.; Shehroz, M.; Hizbullah; Shah, M.; Khan, M.A.; Afridi, S.G.; Khan, A. Potential druggable proteins and chimeric vaccine construct prioritization against Brucella melitensis from species core genome data. Genomics 2020, 112, 1734–1745. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2012, F, 684–687. [Google Scholar] [CrossRef]
- Badhy, S.C.; Chowdhury, M.G.A.; Settypalli, T.B.K.; Cattoli, G.; Lamien, C.E.; Fakir, M.A.U.; Akter, S.; Osmani, M.G.; Talukdar, F.; Begum, N.; et al. Molecular characterization of lumpy skin disease virus (LSDV) emerged in Bangladesh reveals unique genetic features compared to contemporary field strains. BMC Vet. Res. 2021, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2020, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Almehmadi, M.; Allahyani, M.; Alsaiari, A.A.; Alshammari, M.K.; Alharbi, A.S.; Hussain, K.H.; Alsubaihi, L.I.; Kamal, M.; Alotaibi, S.S.; Alotaibi, A.N.; et al. A Glance at the Development and Patent Literature of Tecovirimat: The First-in-Class Therapy for Emerging Monkeypox Outbreak. Viruses 2022, 14, 1870. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Vitalis, T.Z.; Zhang, Q.-J.; Alimonti, J.B.; Chen, S.S.-P.; Basha, G.; Choi, K.B. Pox Viridae Treatment. US7976850B2, 12 July 2011. [Google Scholar]
- Ghosh, P.; Bhattacharya, M.; Patra, P.; Sharma, G.; Patra, B.C.; Lee, S.-S.; Sharma, A.R.; Chakraborty, C. Evaluation and Designing of Epitopic-Peptide Vaccine Against Bunyamwera orthobunyavirus Using M-Polyprotein Target Sequences. Int. J. Pept. Res. Ther. 2021, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Oyarzun, P.; Kobe, B. Computer-aided design of T-cell epitope-based vaccines: Addressing population coverage. Int. J. Immunogenet. 2015, 42, 313–321. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
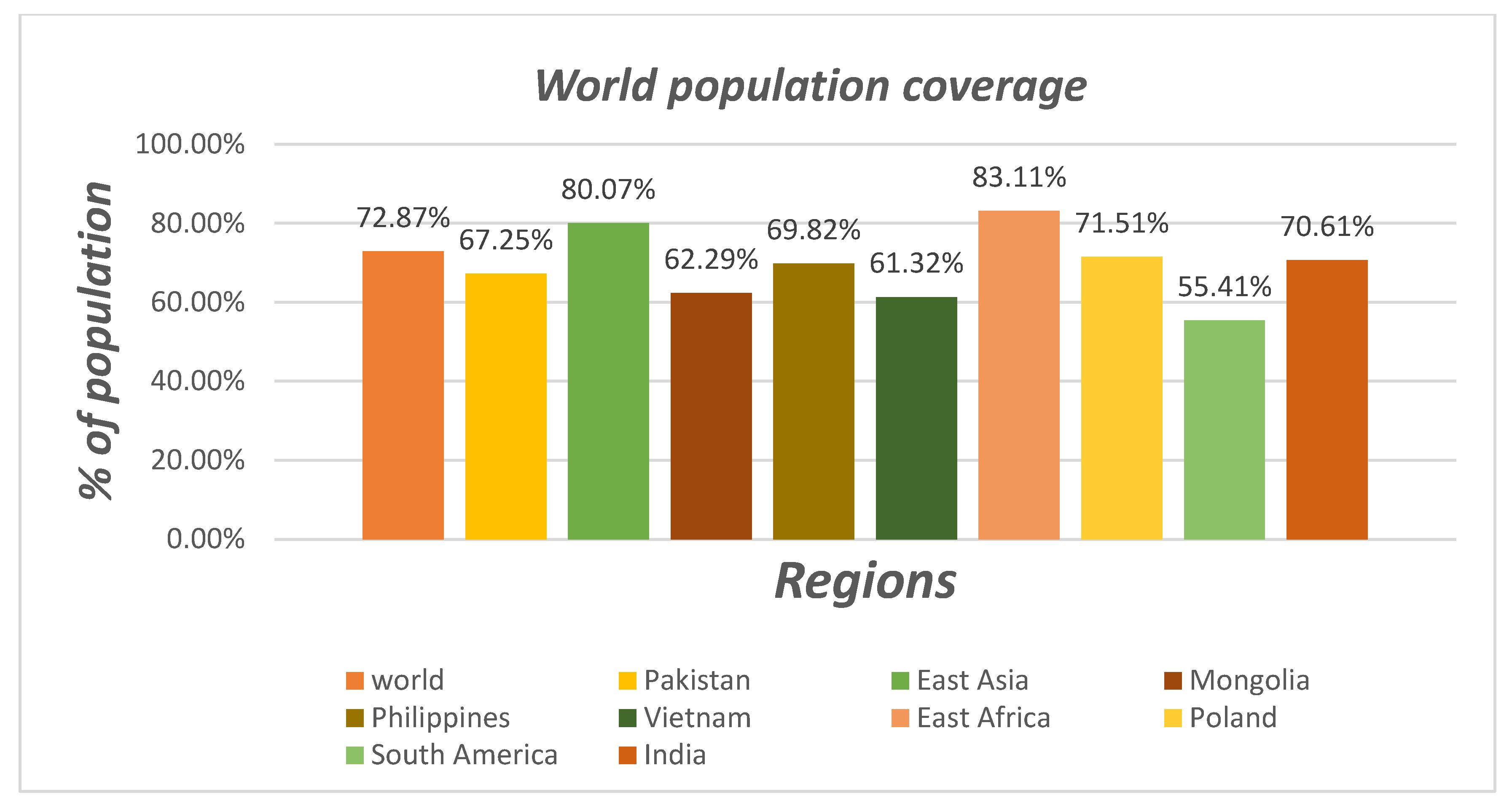{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Protein Name | Sequence | VaxiJen Score | Antigenicity |
|---|---|---|---|---|
| QIN91633.1 | putative virion core protein | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKPNKKSKSCSNKQTTSRSGNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5504 | ANTIGEN |
| AAN02671.1 | putative virion core protein [Lumpy skin disease virus NW-LW] | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKPNKKSKSCSNKQTTSRSGNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5504 | ANTIGEN |
| NP_150537.1 | LSDV103 putative virion core protein | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKPNKKSKSCSNKQTTSRSSNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5529 | ANTIGEN |
| QZZ09333.1 | putative virion core protein | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKPNKKSKSCSNKQTTSRSSNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVKTADDSIDDGMD | 0.5677 | ANTIGEN |
| QBB72816.1 | putative virion core protein, partial | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKLNKKSKSCSNKQTTSRSGNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5513 | ANTIGEN |
| QNN94414.1 | putative virion core protein | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTNNNTDENEVKAGNVKTKACMTKPNKKSKSCS NKQTTSRSSNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRN KNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5433 | ANTIGEN |
| YP_001293294.1 | hypothetical protein GTPV_gp099 [Goat pox virus Pellor] | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTNNNTDENEVKAGNVKTKACITKPNKKSKSCSNKQTTSRSGNVCSSKSVNNGSVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5276 | ANTIGEN |
| AAN02828.1 | putative virion core protein | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTNNNTDENEVKAGNVKTKACMTKTNKKSKSCSNKQTTSRSGNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5508 | ANTIGEN |
| ASF87551.1 | virion core protein [Goat pox virus] | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTNNNTDENEVRAGNVKTKACITKPNKKSKSCSNKQTTSRSGNVCSSKSVNNGSVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5260 | ANTIGEN |
| QEJ79251.1 | core protein [Goat pox virus] | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQTSQKSNLTPEDNTTNNTDENEVKAGNVKTKACITKPNKKSKSCSNKQTTSKSGNVCSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETTDDSIDDGMD | 0.5648 | ANTIGEN |
| NP_659675.1 | Virion core protein [Sheep pox virus] | MSDKKLSRSSYDDYIETINKLTPQLRTILAHISGEQASQKSNLTPEDNTTNNIDENEVKAGNVKTKTCITKPNKKSKSCSNKQTTSRSGNVSSSKSVNNGAVFKKRNTFNETDQIMQAVTNGGKIVYGTMKEGKLEVQGMVGEINQDLLGIESVNAGRRNKNISQSKKKLIKRGMYKVETADDSIDDGMD | 0.5394 | ANTIGEN |
| Start | End | Peptide | Length |
|---|---|---|---|
| 33 | 72 | SGEQASQKSNLTPEDNTTNNTDENEVKAGNVKTKACMTKP | 39 |
| 80 | 121 | NKKSKSCSNKQTTSRSSNVCSSKSVNNGAVFKKRNTFNETDQ | 41 |
| 153 | 187 | SVNAGRRNKNISQSKKKLIKRGMYKVKTADDSIDD | 35 |
| Start | End | Peptide | Antigenic Score | Length |
|---|---|---|---|---|
| 4 | 13 | YGTMKEGKLE | 0.5095 | 10 |
| 70 | 78 | ACMTKPNKK | 0.6486 | 9 |
| 54 | 63 | ACMTKPNKKS | 0.5623 | 10 |
| 71 | 79 | AVTNGGKIV | 0.4661 | 9 |
| 44 | 52 | CMTKPNKKS | 0.9822 | 9 |
| Start | End | Peptide | Antigenic Score | Length |
|---|---|---|---|---|
| 18 | 26 | LTPQLRTIL | 0.6581 | 9 |
| 64 | 72 | LLGIESVNA | 1.4577 | 9 |
| 7 | 15 | KRGMYKVKT | 0.4977 | 9 |
| 61 | 69 | ACMTKPNKK | 0.6486 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahab, M.; Alzahrani, A.K.; Duan, X.; Aslam, M.; Abida; Imran, M.; Kamal, M.; Alam, M.T.; Zheng, G. An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease. Biomedicines 2023, 11, 398. https://doi.org/10.3390/biomedicines11020398
Shahab M, Alzahrani AK, Duan X, Aslam M, Abida, Imran M, Kamal M, Alam MT, Zheng G. An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease. Biomedicines. 2023; 11(2):398. https://doi.org/10.3390/biomedicines11020398
Chicago/Turabian StyleShahab, Muhammad, A. Khuzaim Alzahrani, Xiuyuan Duan, Muneeba Aslam, Abida, Mohd. Imran, Mehnaz Kamal, Md. Tauquir Alam, and Guojun Zheng. 2023. "An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease" Biomedicines 11, no. 2: 398. https://doi.org/10.3390/biomedicines11020398
APA StyleShahab, M., Alzahrani, A. K., Duan, X., Aslam, M., Abida, Imran, M., Kamal, M., Alam, M. T., & Zheng, G. (2023). An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease. Biomedicines, 11(2), 398. https://doi.org/10.3390/biomedicines11020398