
Towards Quantum-Chemical Level Calculations of SARS-CoV-2 Spike Protein Variants of Concern by First Principles Density Functional Theory


Abstract
:1. Introduction
2. Computational Models and Methods
2.1. Modelling Structures from Various PDBs
2.2. Ab Initio Computational Packages
2.3. Key Concepts AABP and AABPU
3. Results
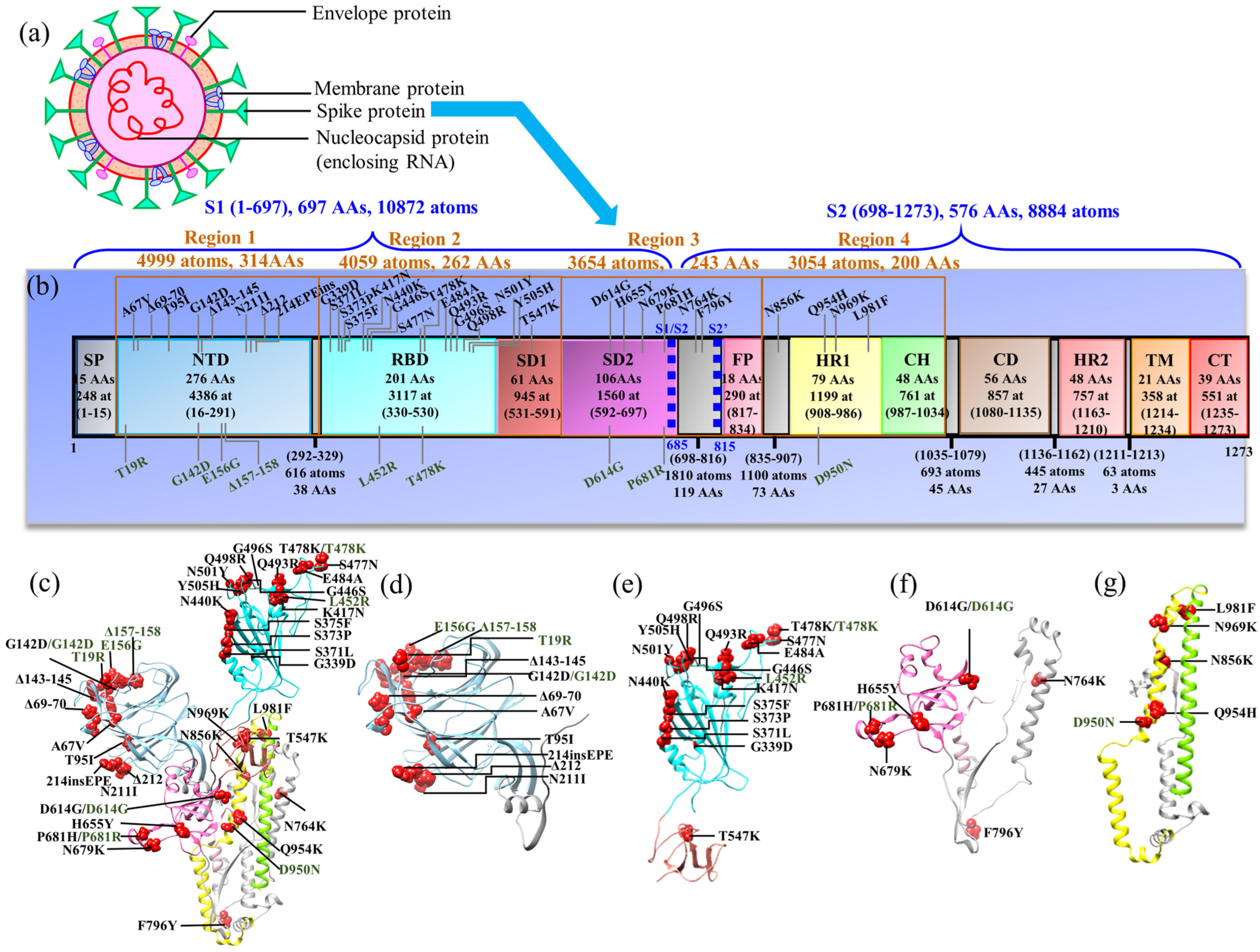
3.1. Structure Domains in Four Regions of S-protein (NTD, RBD–SD1, SD2–FP, and HR1–CH)
3.2. RBD–ACE2 Interface Complex
3.3. Challenges and Opportunities in First Principles DFT Calculation for SARS-CoV-2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 17 December 2022).
- Kupferschmidt, K. New mutations raise specter of ‘immune escape’. Science 2021, 371, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Božič, A.; Kanduč, M. Relative humidity in droplet and airborne transmission of disease. J. Biol. Phys. 2021, 47, 1–29. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Tian, J.-H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef]
- Kang, Y.-F.; Sun, C.; Zhuang, Z.; Yuan, R.-Y.; Zheng, Q.; Li, J.-P.; Zhou, P.-P.; Chen, X.-C.; Liu, Z.; Zhang, X.; et al. Rapid Development of SARS-CoV-2 Spike Protein Receptor-Binding Domain Self-Assembled Nanoparticle Vaccine Candidates. ACS Nano 2021, 15, 2738–2752. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.-G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ryu, D.-K.; Lee, J.; Kim, Y.-I.; Seo, J.-M.; Kim, Y.-G.; Jeong, J.-H.; Kim, M.; Kim, J.-I.; Kim, P.; et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021, 12, 288. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, B.; Kumari, P.; Kumar, P.V.; Agnihotri, G.; Khan, S.; Kant Beuria, T.; Syed, G.H.; Dixit, A. Identification of multipotent drugs for COVID-19 therapeutics with the evaluation of their SARS-CoV2 inhibitory activity. Comput. Struct. Biotechnol. J. 2021, 19, 1998–2017. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Choi, Y.K.; Cao, Y.; Frank, M.; Woo, H.; Park, S.-J.; Yeom, M.S.; Croll, T.I.; Seok, C.; Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. J. Chem. Theory Comput. 2021, 17, 2479–2487. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Central Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.-H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Brotzakis, Z.F.; Löhr, T.; Vendruscolo, M. Determination of intermediate state structures in the opening pathway of SARS-CoV-2 spike using cryo-electron microscopy. Chem. Sci. 2021, 12, 9168–9175. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Shin, M.; Steinmetz, N.F.; Twarock, R.; Podgornik, R.; Ching, W.-Y. Intra- and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: Implication for ACE2 receptor binding. Phys. Chem. Chem. Phys. 2020, 22, 18272–18283. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Ching, W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020, 10, 39831–39841. [Google Scholar] [CrossRef] [PubMed]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Ultra-large-scale ab initio quantum chemical computation of bio-molecular systems: The case of spike protein of SARS-CoV-2 virus. Comput. Struct. Biotechnol. J. 2021, 19, 1288–1301. [Google Scholar] [CrossRef]
- Adhikari, P.; Podgornik, R.; Jawad, B.; Ching, W.-Y. First-Principles Simulation of Dielectric Function in Biomolecules. Materials 2021, 14, 5774. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Rao, P.; Podgornik, R.; Ching, W.-Y. Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions. Viruses 2022, 14, 465. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Quantum Chemical Computation of Omicron Mutations Near Cleavage Sites of the Spike Protein. Microorganisms 2022, 10, 1999. [Google Scholar] [CrossRef] [PubMed]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Effect of Delta and Omicron Mutations on the RBD-SD1 Domain of the Spike Protein in SARS-CoV-2 and the Omicron Mutations on RBD-ACE2 Interface Complex. Int. J. Mol. Sci. 2022, 23, 10091. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Mutations of Omicron Variant at the Interface of the Receptor Domain Motif and Human Angiotensin-Converting Enzyme-2. Int. J. Mol. Sci. 2022, 23, 2870. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Binding Interactions between Receptor-Binding Domain of Spike Protein and Human Angiotensin Converting Enzyme-2 in Omicron Variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Cheng, K.; Podgornik, R.; Ching, W.-Y. Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors. Int. J. Mol. Sci. 2022, 23, 838. [Google Scholar] [CrossRef]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef]
- Ye, G.; Bin Liu, B.; Li, F. Cryo-EM structure of a SARS-CoV-2 omicron spike protein ectodomain. Nat. Commun. 2022, 13, 1214. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- VASP—Vienna Ab initio Simulation Package. Available online: https://www.vasp.at/ (accessed on 1 November 2021).
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Ching, W.-Y.; San, S.; Brechtl, J.; Sakidja, R.; Zhang, M.; Liaw, P.K. Fundamental electronic structure and multiatomic bonding in 13 biocompatible high-entropy alloys. NPJ Comput. Mater. 2020, 6, 45. [Google Scholar] [CrossRef]
- Ching, W.Y. Ceramic Genome: Total Bond Order Density. In Encyclopedia of Materials: Technical Ceramics and Glasses; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Ching, W.-Y.; Poudel, L.; San, S.; Baral, K. Interfacial Interaction between Suolunite Crystal and Silica Binding Peptide for Novel Bioinspired Cement. ACS Comb. Sci. 2019, 21, 794–804. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Rulis, P.; Ching, W.-Y. Deformation behavior of an amorphous zeolitic imidazolate framework—From a supersoft material to a complex organometallic alloy. Phys. Chem. Chem. Phys. 2018, 20, 29001–29011. [Google Scholar] [CrossRef]
- Baral, K.; Adhikari, P.; Ching, W. Ab initio Modeling of the Electronic Structures and Physical Properties of a-Si1−xGexO2 Glass (x = 0 to 1). J. Am. Ceram. Soc. 2016, 99, 3677–3684. [Google Scholar] [CrossRef]
- Poudel, L.; Rulis, P.; Liang, L.; Ching, W.Y. Electronic structure, stacking energy, partial charge, and hydrogen bonding in four periodic B-DNA models. Phys. Rev. E 2014, 90, 022705. [Google Scholar] [CrossRef] [PubMed]
- San, S.; Ching, W.-Y. Subtle Variations of the Electronic Structure and Mechanical Properties of High Entropy Alloys With 50% Carbon Composites. Front. Mater. 2020, 7, 371. [Google Scholar] [CrossRef]
- Hasan, S.; Baral, K.; Li, N.; Ching, W.-Y. Structural and physical properties of 99 complex bulk chalcogenides crystals using first-principles calculations. Sci. Rep. 2021, 11, 9921. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Rulis, P.; Ching, W.-Y. First-Principles Calculations of the Structural, Electronic, Optical, and Mechanical Properties of 21 Pyrophosphate Crystals. Crystals 2022, 12, 1139. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Meng, B.; Datir, R.; Choi, J.; Bradley, J.R.; Smith, K.G.; Lee, J.H.; Gupta, R.K.; Baker, S.; Dougan, G.; Hess, C.; et al. SARS-CoV-2 spike N-terminal domain modulates TMPRSS2-dependent viral entry and fusogenicity. Cell Rep. 2022, 40, 111220. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, Y.; Zhang, S.; Niu, P.; Qin, K.; Jia, W.; Huang, B.; Zhang, S.; Lan, J.; Zhang, L.; et al. Structural definition of a neutralization epitope on the N-terminal domain of MERS-CoV spike glycoprotein. Nat. Commun. 2019, 10, 3068. [Google Scholar] [CrossRef]
- Qing, E.; Kicmal, T.; Kumar, B.; Hawkins, G.M.; Timm, E.; Perlman, S.; Gallagher, T. Dynamics of SARS-CoV-2 Spike Proteins in Cell Entry: Control Elements in the Amino-Terminal Domains. Mbio 2021, 12, e01590-21. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef]
- Ouyang, W.O.; Tan, T.J.; Lei, R.; Song, G.; Kieffer, C.; Andrabi, R.; Matreyek, K.A.; Wu, N.C. Probing the biophysical constraints of SARS-CoV-2 spike N-terminal domain using deep mutational scanning. Sci. Adv. 2022, 8, eadd7221. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, L.; Segovia-Chumbez, B.; Jadi, R.; Martinez, D.R.; Raut, R.; Markmann, A.; Cornaby, C.; Bartelt, L.; Weiss, S.; Park, Y.; et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci. Immunol. 2020, 5, eabc8413. [Google Scholar] [CrossRef]
- Yang, J.; Wang, W.; Chen, Z.; Lu, S.; Yang, F.; Bi, Z.; Bao, L.; Mo, F.; Li, X.; Huang, Y.; et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 2020, 586, 572–577. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.H.; Bastidas, O.; Kokron, D.S.; Henze, C.E. Static all-atom energetic mappings of the SARS-Cov-2 spike protein and dynamic stability analysis of “Up” versus “Down” protomer states. PLoS ONE 2020, 15, e0241168. [Google Scholar] [CrossRef] [PubMed]
- French, R.H.; Parsegian, V.A.; Podgornik, R.; Rajter, R.F.; Jagota, A.; Luo, J.; Asthagiri, D.; Chaudhury, M.K.; Chiang, Y.-M.; Granick, S.; et al. Long range interactions in nanoscale science. Rev. Mod. Phys. 2010, 82, 1887–1944. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Sarkar, S.; Zhang, J.; Witham, S.; Zhang, Z.; Wang, L.; Smith, N.; Petukh, M.; Alexov, E. DelPhi: A comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, K.; Whiteley, M.; de Silva, T.I.; Barclay, W.S.; Consortium, P.V. The SARS-CoV-2 variants associated with infections in India, B. 1.617, show enhanced spike cleavage by furin. bioRxiv 2021. [Google Scholar] [CrossRef]
- Saito, A.; Nasser, H.; Uriu, K.; Kosugi, Y.; Irie, T.; Shirakawa, K. SARS-CoV-2 spike P681R mutation enhances and accelerates viral fusion. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. Iscience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Raghav, S.; Ghosh, A.; Turuk, J.; Kumar, S.; Jha, A.; Madhulika, S.; Priyadarshini, M.; Biswas, V.K.; Shyamli, P.S.; Singh, B.; et al. Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614G Mutation in Spike Protein Predicting an Increase in Interaction with TMPRSS2 and Virus Infectivity. Front. Microbiol. 2020, 11, 594928. [Google Scholar] [CrossRef]
- Lubinski, B.; Jaimes, J.A.; Whittaker, G.R. Intrinsic furin-mediated cleavability of the spike S1/S2 site from SARS-CoV-2 variant B. 1.529 (Omicron). bioRxiv 2022. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Du, X.; Tang, H.; Gao, L.; Wu, Z.; Meng, F.; Yan, R.; Qiao, S.; An, J.; Wang, C.; Qin, F.X.-F. Omicron adopts a different strategy from Delta and other variants to adapt to host. Signal Transduct. Target. Ther. 2022, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Ng, O.-W.; Keng, C.-T.; Leung, C.S.-W.; Peiris, J.S.M.; Poon, L.L.M.; Tan, Y.-J. Substitution at Aspartic Acid 1128 in the SARS Coronavirus Spike Glycoprotein Mediates Escape from a S2 Domain-Targeting Neutralizing Monoclonal Antibody. PLoS ONE 2014, 9, e102415. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Coughlin, M.M.; Baker, S.C.; Prabhakar, B.S. Human Monoclonal Antibodies against Highly Conserved HR1 and HR2 Domains of the SARS-CoV Spike Protein Are More Broadly Neutralizing. PLoS ONE 2012, 7, e50366. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhu, J.; Zhang, J.; Yan, M.; Gao, G.F.; Tien, P. Design of recombinant protein-based SARS-CoV entry inhibitors targeting the heptad-repeat regions of the spike protein S2 domain. Biochem. Biophys. Res. Commun. 2005, 330, 39–45. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19′s next move. Cell Host Microbe. 2021, 29, 508–515. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based COVID-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; He, X.; Ren, Y.; Wang, Z.; Zhou, H.; Fan, S.; Zhu, C.; Liu, D.; Bin Shao, B.; Liu, T.-Y.; et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022, 32, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Impact of BA.1, BA.2, and BA.4/BA.5 Omicron Mutations on Therapeutic Monoclonal Antibodies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Watanabe, C.; Okiyama, Y.; Tanaka, S.; Fukuzawa, K.; Honma, T. Molecular recognition of SARS-CoV-2 spike glycoprotein: Quantum chemical hot spot and epitope analyses. Chem. Sci. 2021, 12, 4722–4739. [Google Scholar] [CrossRef]
- Lim, H.; Baek, A.; Kim, J.; Kim, M.S.; Liu, J.; Nam, K.-Y.; Yoon, J.; No, K.T. Hot spot profiles of SARS-CoV-2 and human ACE2 receptor protein protein interaction obtained by density functional tight binding fragment molecular orbital method. Sci. Rep. 2020, 10, 16862. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, C.; Honma, T.; Tian, Y.-S.; Kawashima, Y.; Kawashita, N.; Takagi, T.; Fukuzawa, K. Intermolecular Interaction Analyses on SARS-CoV-2 Spike Protein Receptor Binding Domain and Human Angiotensin-Converting Enzyme 2 Receptor-Blocking Antibody/Peptide Using Fragment Molecular Orbital Calculation. J. Phys. Chem. Lett. 2021, 12, 4059–4066. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Total AABP | NN AABP | Non-local AABP | AABP from HB | No. of NL AAs |
|---|---|---|---|---|---|
| WT T19 | 1.121 | 1.053 | 0.068 | 0.124 | 1 |
| DV R19 | 0.959 | 0.958 | 0.001 | 0.024 | 3 |
| WT G142 | 1.147 | 1.014 | 0.133 | 0.154 | 10 |
| DV D142 | 1.030 | 0.940 | 0.090 | 0.100 | 4 |
| WT E156 | 1.208 | 0.959 | 0.249 | 0.229 | 10 |
| DV G156 | 0.918 | 0.465 | 0.453 | 0.029 | 4 |
| WT A67 | 1.064 | 0.989 | 0.075 | 0.092 | 7 |
| OV V67 | 1.046 | 0.965 | 0.081 | 0.093 | 10 |
| WT T95 | 1.053 | 0.998 | 0.055 | 0.075 | 9 |
| OV I95 | 1.079 | 1.008 | 0.072 | 0.084 | 11 |
| WT G142 | 1.147 | 1.014 | 0.133 | 0.154 | 10 |
| OV D142 | 1.265 | 1.007 | 0.257 | 0.251 | 9 |
| WT N211 | 1.028 | 1.028 | 0.031 | 0.089 | 3 |
| OV I211 | 0.949 | 0.944 | 0.005 | 0.031 | 5 |
| Models | Total AABP | NN AABP | NL AABP | No. of HBs (HB AABP) | No. of NL AAs | Volume (Å3) | Area (Å2) | PC* (e−) |
|---|---|---|---|---|---|---|---|---|
| WT L452 | 1.022 | 0.978 | 0.045 | 31 (0.061) | 9 | 1641.0 | 1048.0 | −0.074 |
| DV R452 | 1.019 | 0.976 | 0.043 | 26 (0.064) | 8 | 1549.0 | 972.8 | 0.849 |
| WT T478 | 1.044 | 1.043 | 0.001 | 9 (0.022) | 1 | 335.1 | 333.5 | 0.005 |
| DV K478 | 1.219 | 1.217 | 0.002 | 13 (0.136) | 3 | 571.1 | 497.8 | 1.022 |
| WT G339 | 1.016 | 0.993 | 0.023 | 11 (0.052) | 3 | 652.2 | 570.6 | −0.340 |
| OV D339 | 1.196 | 1.154 | 0.042 | 14 (0.063) | 4 | 807.7 | 634.1 | −1.357 |
| WT S371 | 0.918 | 0.888 | 0.030 | 20 (0.051) | 5 | 854.7 | 680.5 | −0.147 |
| OV L371 | 0.945 | 0.928 | 0.017 | 21 (0.040) | 3 | 608.7 | 532.5 | −0.162 |
| WT S373 | 0.941 | 0.920 | 0.021 | 14 (0.052) | 3 | 633.6 | 543.4 | −0.075 |
| OV P373 | 0.999 | 0.992 | 0.008 | 16 (0.031) | 3 | 764.9 | 623.2 | −0.084 |
| WT S375 | 0.944 | 0.916 | 0.028 | 11 (0.058) | 4 | 808.4 | 642.2 | −0.026 |
| OV F375 | 0.926 | 0.917 | 0.009 | 13 (0.037) | 7 | 1331.0 | 941.1 | 0.076 |
| WT K417 | 1.216 | 1.013 | 0.203 | 19 (0.203) | 7 | 1195.0 | 827.5 | 0.153 |
| OV N417 | 1.066 | 1.017 | 0.048 | 14 (0.069) | 6 | 987.4 | 697.0 | −1.473 |
| WT N440 | 0.985 | 0.981 | 0.005 | 12 (0.037) | 3 | 584.2 | 467.3 | −0.802 |
| OV K440 | 0.983 | 0.978 | 0.005 | 14 (0.037) | 4 | 825.4 | 645.6 | 0.148 |
| WT G446 | 0.912 | 0.910 | 0.002 | 10 (0.038) | 2 | 473.8 | 403.3 | 0.907 |
| OV S446 | 1.038 | 0.979 | 0.059 | 14 (0.091) | 2 | 530.4 | 443.5 | 1.843 |
| WT S477 | 0.964 | 0.958 | 0.006 | 12 (0.039) | 2 | 440.7 | 383.3 | 0.100 |
| OV N477 | 1.157 | 1.156 | 0.001 | 11 (0.151) | 2 | 507.3 | 428.0 | 1.097 |
| WT T478 | 1.044 | 1.043 | 0.001 | 9 (0.022) | 1 | 335.1 | 333.5 | 0.005 |
| OV K478 | 1.214 | 1.212 | 0.002 | 13 (0.139) | 3 | 594.9 | 509.1 | 1.045 |
| WT E484 | 1.040 | 0.927 | 0.114 | 19 (0.124) | 4 | 828.5 | 633.4 | −0.967 |
| OV A484 | 0.934 | 0.932 | 0.002 | 13 (0.030) | 3 | 513.8 | 452.9 | −0.081 |
| WT Q493 | 1.060 | 0.973 | 0.087 | 19 (0.106) | 6 | 1220.0 | 786.3 | 0.497 |
| OV R493 | 1.165 | 1.165 | 0.194 | 32 (0.200) | 9 | 1739.0 | 1034.0 | −0.498 |
| WT G496 | 0.975 | 0.944 | 0.031 | 11 (0.062) | 4 | 834.4 | 691.4 | −0.285 |
| OV S496 | 0.994 | 0.938 | 0.055 | 12 (0.076) | 4 | 964.4 | 755.0 | 0.657 |
| WT Q498 | 1.120 | 1.073 | 0.047 | 25 (0.054) | 10 | 1376.0 | 894.1 | 1.013 |
| OV R498 | 1.179 | 1.056 | 0.123 | 30 (0.126) | 11 | 1648.0 | 1078.0 | 2.059 |
| WT N501 | 1.120 | 1.073 | 0.047 | 27 (0.054) | 10 | 1063.0 | 802.3 | 0.022 |
| OV Y501 | 1.034 | 0.942 | 0.092 | 21 (0.104) | 6 | 1089.0 | 797.4 | 0.752 |
| WT Y505 | 1.058 | 0.974 | 0.084 | 17 (0.104) | 6 | 983.1 | 714.7 | 0.156 |
| OV H505 | 0.998 | 0.953 | 0.045 | 15 (0.069) | 7 | 1188.0 | 859.7 | 0.283 |
| WT T547 | 1.033 | 0.977 | 0.056 | 20 (0.079) | 4 | 738.6 | 527.8 | 0.150 |
| OV K547 | 0.994 | 0.977 | 0.016 | 21 (0.042) | 4 | 773.5 | 584.4 | 1.100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Towards Quantum-Chemical Level Calculations of SARS-CoV-2 Spike Protein Variants of Concern by First Principles Density Functional Theory. Biomedicines 2023, 11, 517. https://doi.org/10.3390/biomedicines11020517
Ching W-Y, Adhikari P, Jawad B, Podgornik R. Towards Quantum-Chemical Level Calculations of SARS-CoV-2 Spike Protein Variants of Concern by First Principles Density Functional Theory. Biomedicines. 2023; 11(2):517. https://doi.org/10.3390/biomedicines11020517
Chicago/Turabian StyleChing, Wai-Yim, Puja Adhikari, Bahaa Jawad, and Rudolf Podgornik. 2023. "Towards Quantum-Chemical Level Calculations of SARS-CoV-2 Spike Protein Variants of Concern by First Principles Density Functional Theory" Biomedicines 11, no. 2: 517. https://doi.org/10.3390/biomedicines11020517
APA StyleChing, W. -Y., Adhikari, P., Jawad, B., & Podgornik, R. (2023). Towards Quantum-Chemical Level Calculations of SARS-CoV-2 Spike Protein Variants of Concern by First Principles Density Functional Theory. Biomedicines, 11(2), 517. https://doi.org/10.3390/biomedicines11020517