

Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Equipment
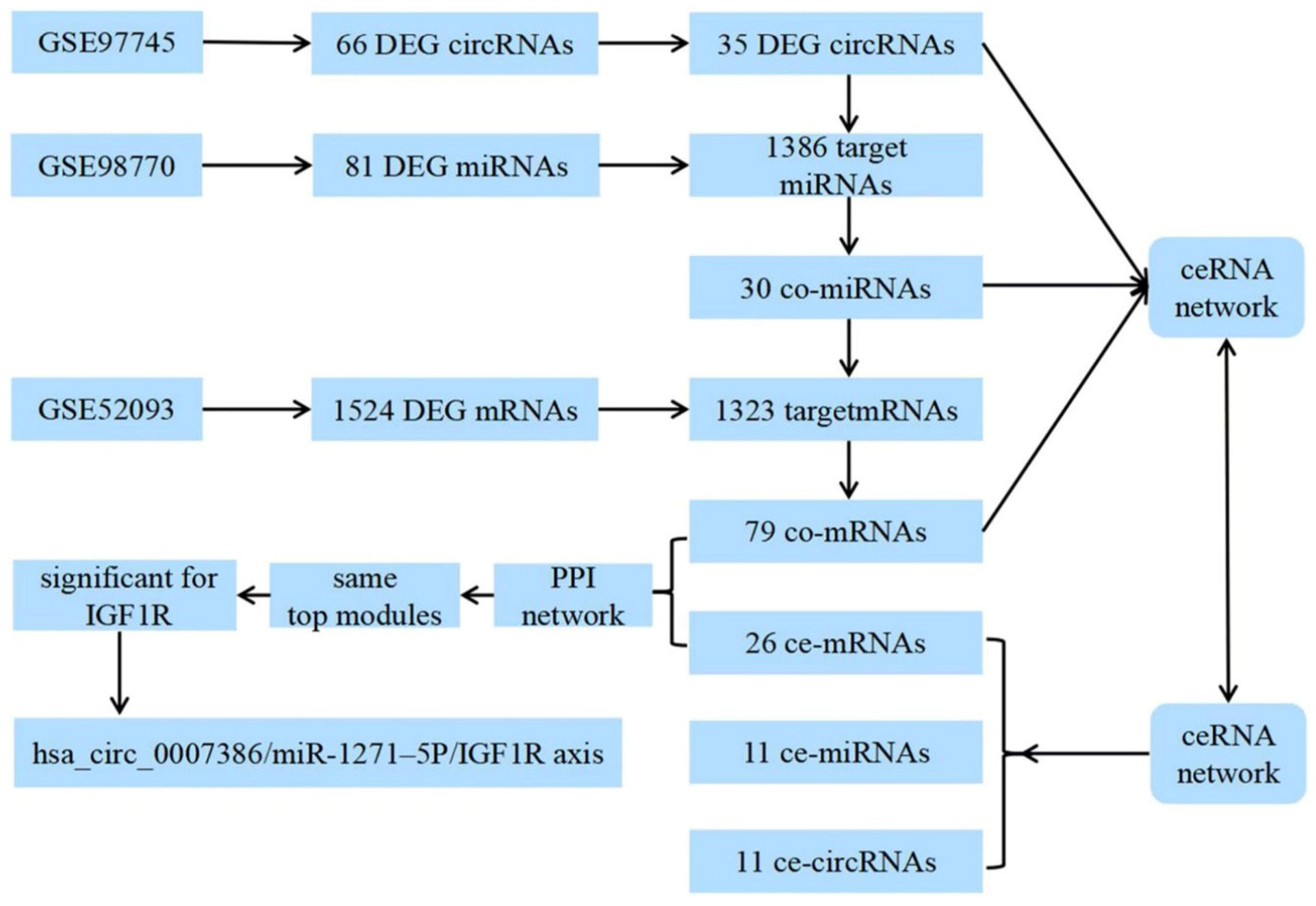
2.2. Datasets and Analysis of Differentially Expressed Genes
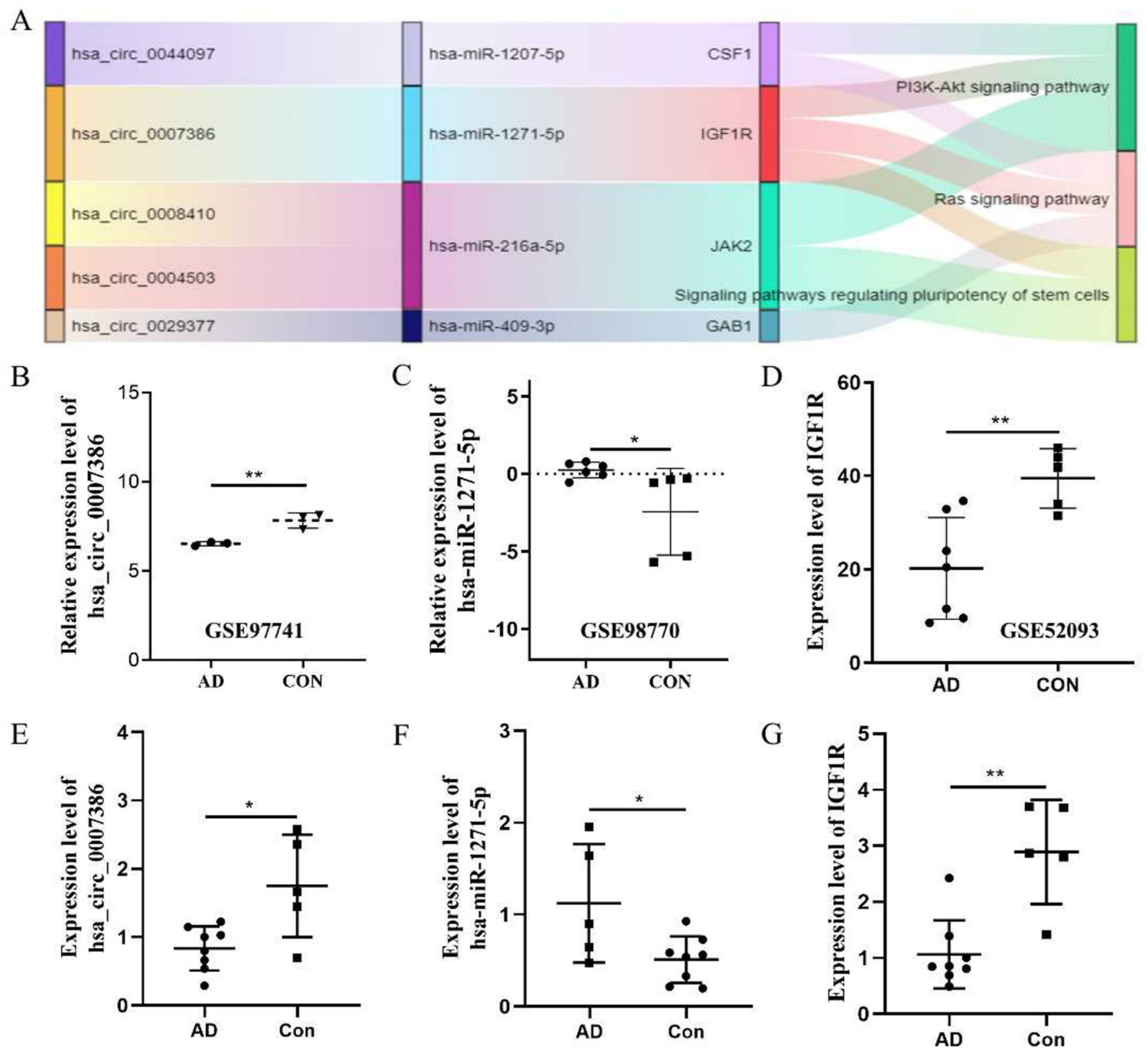
2.3. Construction of the circRNA–microRNA–mRNA Network
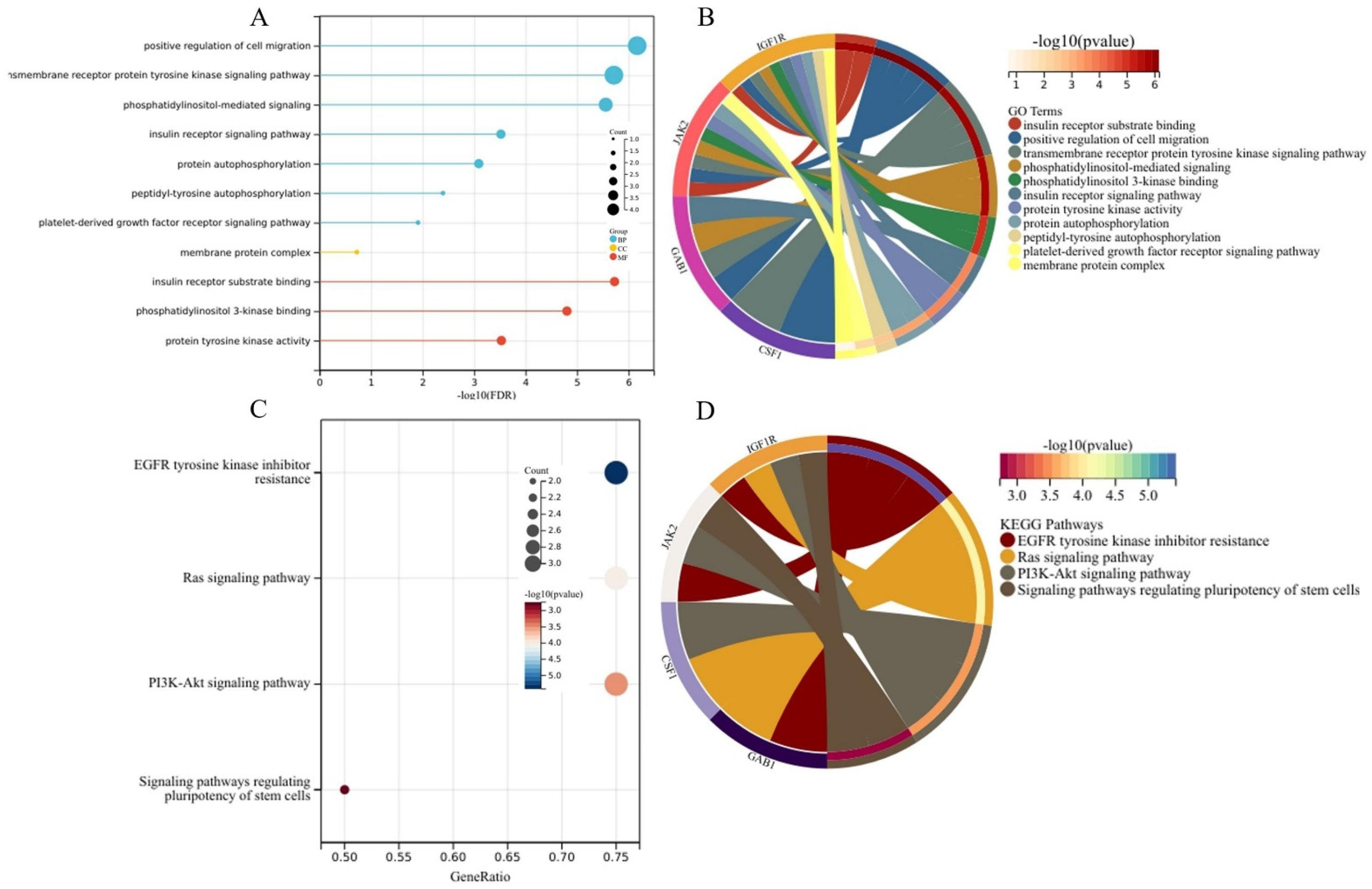
2.4. Generation of the PPI Network and the Functional Enrichment Analysis of Target Genes
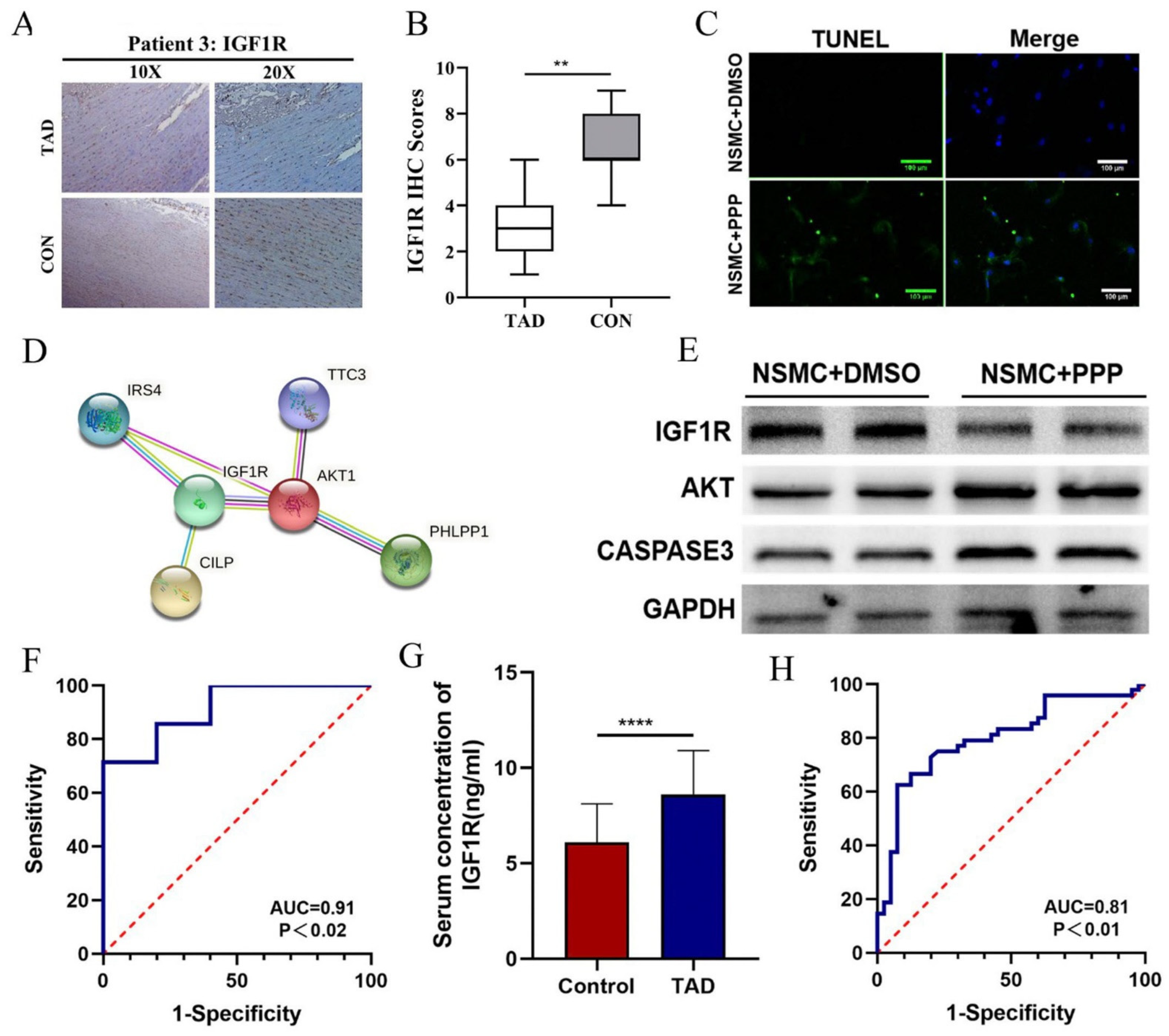
2.5. Immunohistochemistry (IHC), TUNEL Staining and Western Blot Assay
2.6. Statistical Analysis
3. Results
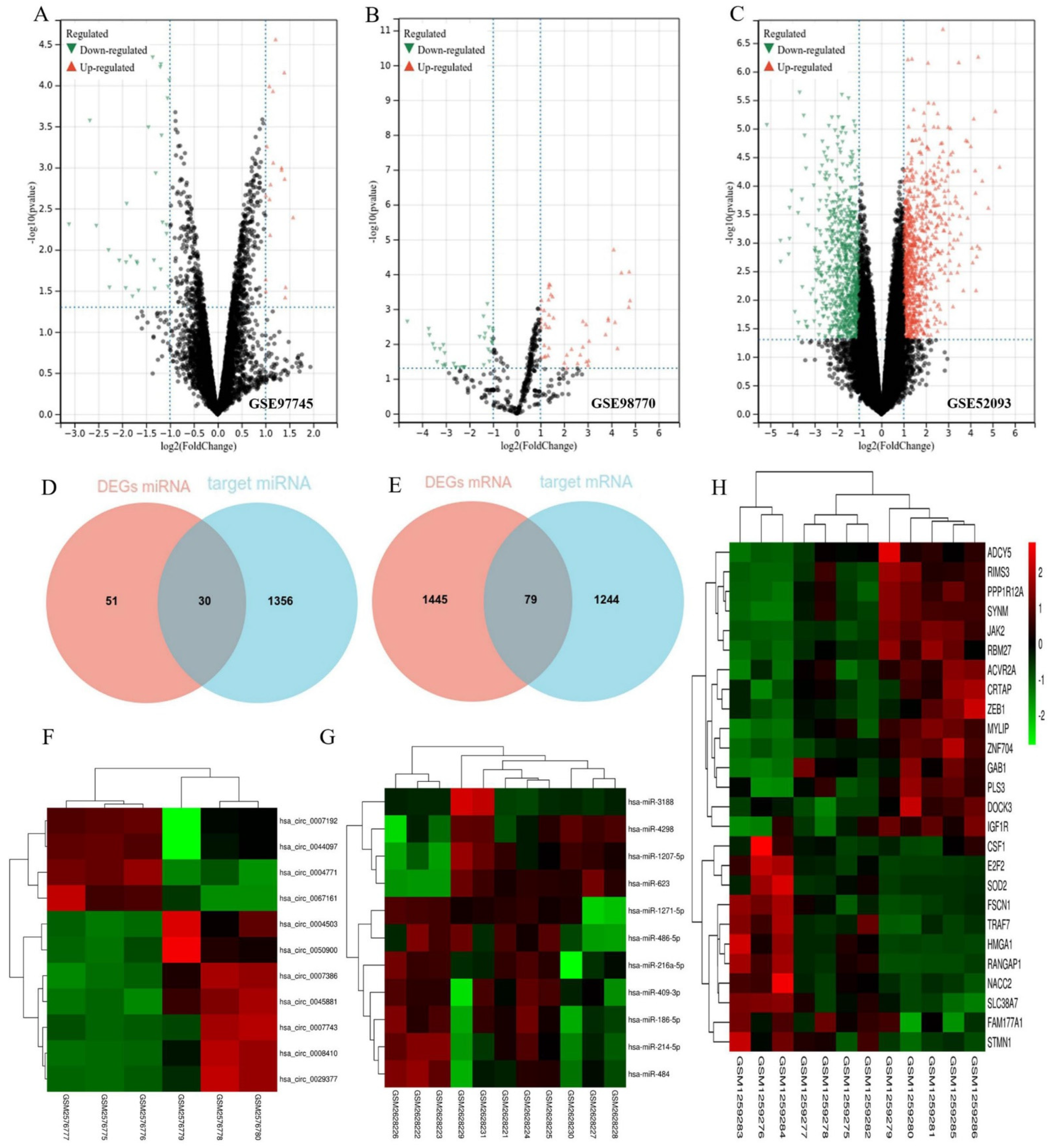
3.1. Identification of DEGs and Target Genes
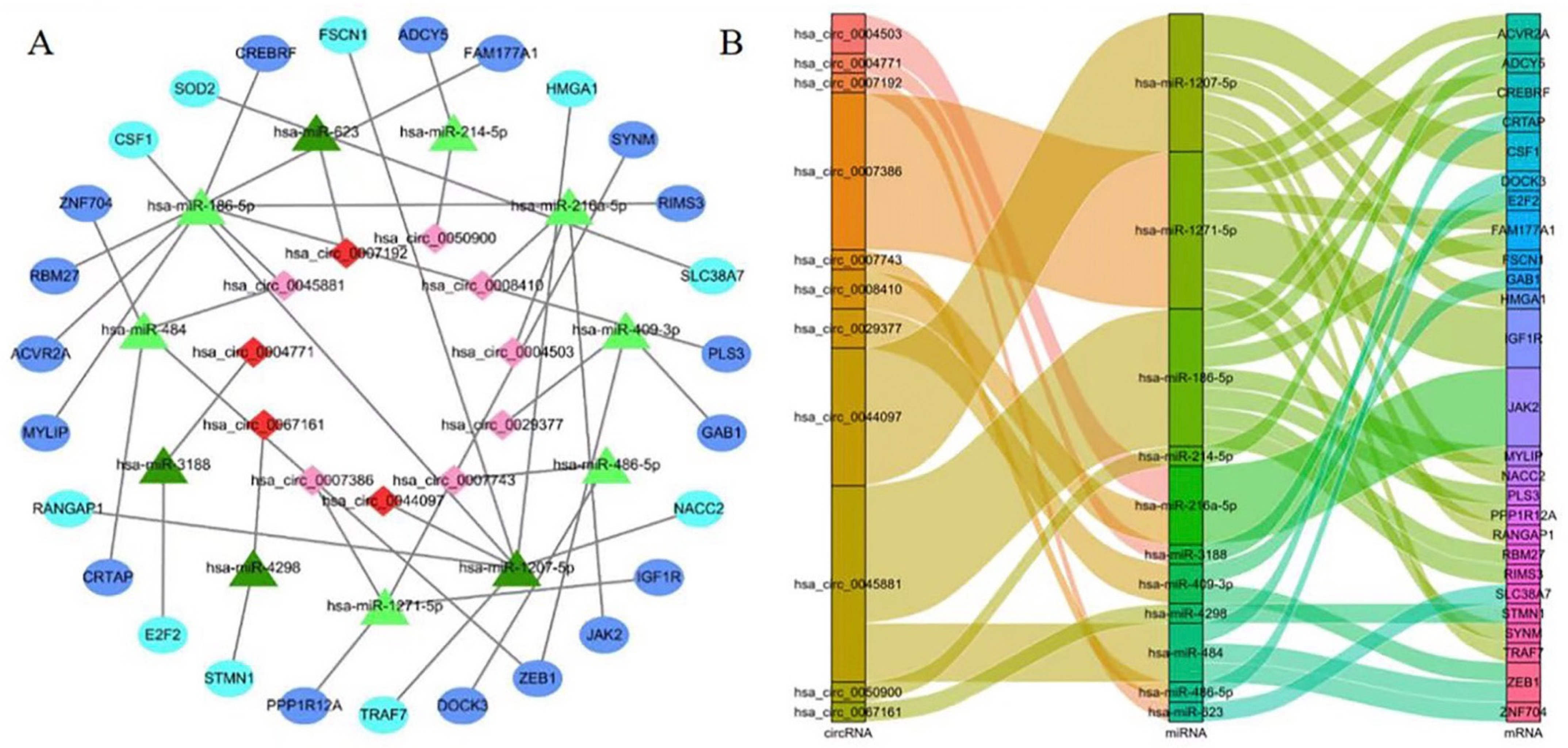
3.2. Construction of the ceRNA Network
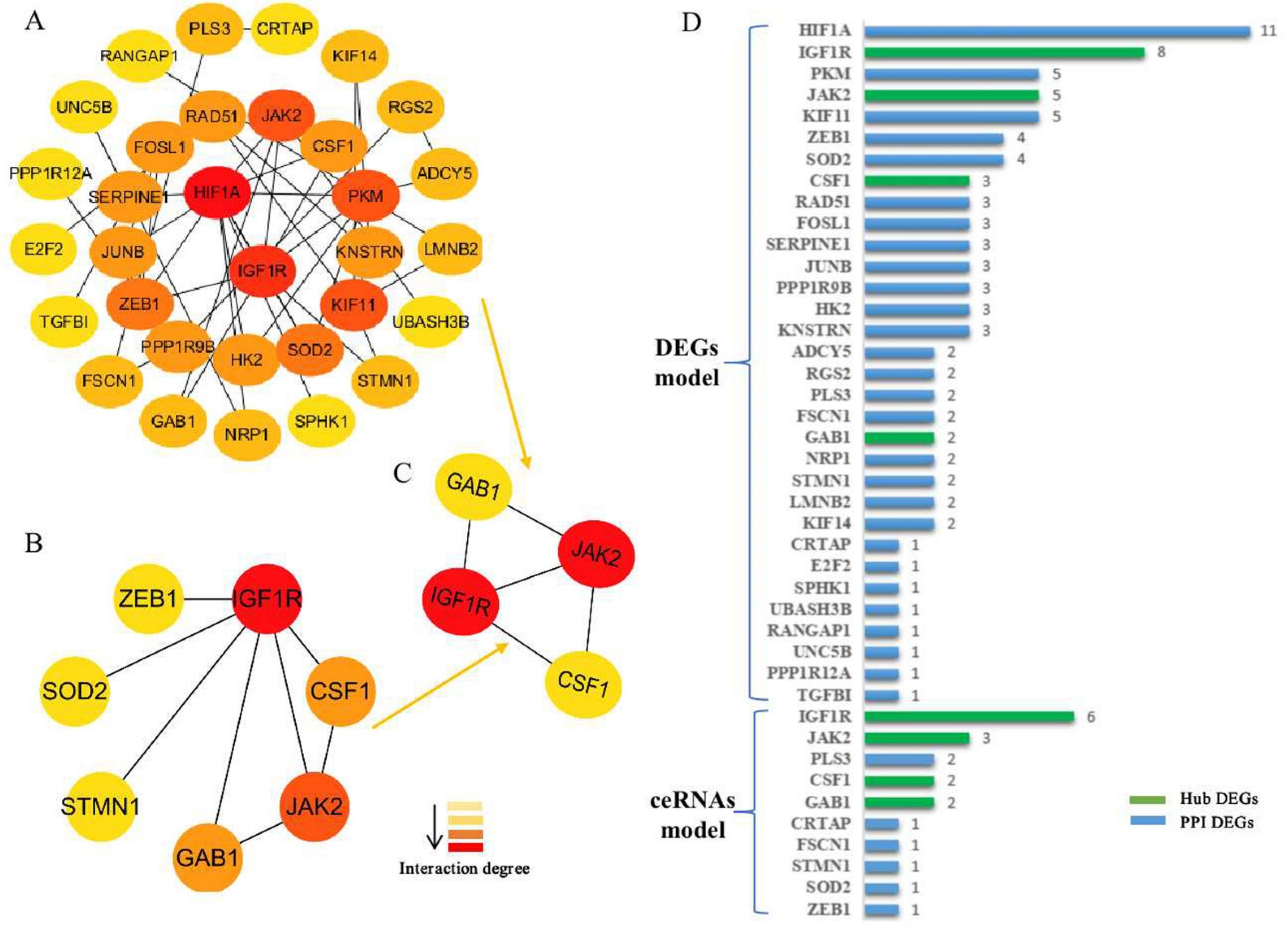
3.3. Generation of the PPI Network and Enrichment Analysis of the Key Module
3.4. The hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis May Aggravate the Progression of TAD by Inducing VSMCs Apoptosis
3.5. IGF1R Can Be Considered as a Diagnostic Marker for TAD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tchana-Sato, V.; Sakalihasan, N.; Defraigne, J. Aortic dissection. Rev. Med. Liege 2018, 73, 290–295. [Google Scholar] [PubMed]
- Delafontaine, P.; Song, Y.; Li, Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arter. Thromb. Vasc. Biol. 2004, 24, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Silaschi, M.; Byrne, J.; Wendler, O. Aortic dissection: Medical, interventional and surgical management. Heart 2017, 103, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Boukhris, M. Dissecting the epidemiology of aortic dissection. Eur. Heart J. Acute Cardiovasc Care 2021, 10, 710–711. [Google Scholar] [CrossRef]
- Parve, S.; A Ziganshin, B.; A Elefteriades, J. Overview of the current knowledge on etiology, natural history and treatment of aortic dissection. J. Cardiovasc Surg. 2017, 58, 238–251. [Google Scholar] [CrossRef]
- Cossu, A.M.; Mosca, L.; Zappavigna, S.; Misso, G.; Bocchetti, M.; De Micco, F.; Quagliuolo, L.; Porcelli, M.; Caraglia, M.; Boccellino, M. Long Non-coding RNAs as Important Biomarkers in Laryngeal Cancer and Other Head and Neck Tumours. Int. J. Mol. Sci. 2019, 20, 3444. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.F. Non-coding RNAs: Meet thy masters. Bioessays 2010, 32, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Han, M.; Wei, G.; Ni, T. An intriguing RNA species—Perspectives of circularized RNA. Protein Cell 2015, 6, 871–880. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Tehrani, S.S.; Ebrahimi, R.; Al-E-Ahmad, A.; Panahi, G.; Meshkani, R.; Younesi, S.; Saadat, P.; Parsian, H. Competing Endogenous RNAs (CeRNAs): Novel Network in Neurological Disorders. Curr. Med. Chem. 2021, 28, 5983–6010. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Jing, Q.; Huang, S.; Guth, S.; Zarubin, T.; Motoyama, A.; Chen, J.; Di Padova, F.; Lin, S.; Gram, H.; Han, J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 2005, 120, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-X.; Xie, X.-S.; Weng, X.-F.; Zheng, C.-H.; Xie, J.-W.; Wang, J.-B.; Lu, J.; Chen, Q.-Y.; Cao, L.-L.; Lin, M.; et al. The prognostic value of Cyclin-Dependent Kinase 5 and Protein Phosphatase 2A in Gastric Cancer. J. Cancer 2018, 9, 4404–4412. [Google Scholar] [CrossRef]
- Lin, J.; Xie, X.; Weng, X.; Qiu, S.; Yoon, C.; Lian, N.; Xie, J.; Wang, J.; Lu, J.; Chen, Q.; et al. UFM1 suppresses invasive activities of gastric cancer cells by attenuating the expres7sion of PDK1 through PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2019, 38, 410. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.; Nastou, K.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [Green Version]
- Bi, S.; Liu, R.; Shen, Y.; Gu, J. Bioinformatics analysis of key genes and miRNAs associated with Stanford type A aortic dissection. J. Thorac. Dis. 2020, 12, 4842–4853. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, Y.; Chen, G.; Huang, H.; Xie, X.; Lin, W.; Peng, Z. Construction of a circRNA-Mediated ceRNA Network Reveals Novel Biomarkers for Aortic Dissection. Int. J. Gen. Med. 2022, 15, 3951–3964. [Google Scholar] [CrossRef]
- Zou, M.; Huang, C.; Li, X.; He, X.; Chen, Y.; Liao, W.; Liao, Y.; Sun, J.; Liu, Z.; Zhong, L.; et al. Circular RNA expression profile and potential function of hsa_circRNA_101238 in human thoracic aortic dissection. Oncotarget 2017, 8, 81825–81837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cheng, H.; Yue, Y.; Li, S.; Zhang, D.; He, R. H19 knockdown suppresses proliferation and induces apoptosis by regulating miR-148b/WNT/β-catenin in ox-LDL -stimulated vascular smooth muscle cells. J. Biomed. Sci. 2018, 25, 11. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Zhao, X.; Cao, L.; Karnad, A.; Kumar, A.; Freeman, J. Gemcitabine resistance of pancreatic cancer cells is mediated by IGF1R dependent upregulation of CD44 expression and isoform switching. Cell Death Dis. 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Seccareccia, E.; Brodt, P. The role of the insulin-like growth factor-I receptor in malignancy: An update. Growth Horm. IGF Res. 2012, 22, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.M.; Bennett, M.R. Expression, regulation and function of trail in atherosclerosis. Biochem. Pharmacol. 2008, 75, 1441–1450. [Google Scholar] [CrossRef]
- Okura, Y.; Brink, M.; Itabe, H.; Scheidegger, K.J.; Kalangos, A.; Delafontaine, P. Oxidized Low-Density Lipoprotein Is Associated With Apoptosis of Vascular Smooth Muscle Cells in Human Atherosclerotic Plaques. Circulation 2000, 102, 2680–2686. [Google Scholar] [CrossRef]
- Du, J.; Delafontaine, P. Inhibition of Vascular Smooth Muscle Cell Growth Through Antisense Transcription of a Rat Insulin-Like Growth Factor I Receptor cDNA. Circ. Res. 1995, 76, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, S.; Higashi, Y.; Shai, S.-Y.; Snarski, P.; Danchuk, S.; D’Ambra, V.; Tabony, M.; Woods, T.C.; Hou, X.; Li, Z.; et al. SM22α (Smooth Muscle Protein 22-α) Promoter-Driven IGF1R (Insulin-Like Growth Factor 1 Receptor) Deficiency Promotes Atherosclerosis. Arter. Thromb. Vasc. Biol. 2018, 38, 2306–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolo, B.; Schoppet, M.; Mattar, M.; Rachner, T.; Shanahan, C.; Kavurma, M. Calcium and osteoprotegerin regulate IGF1R expression to inhibit vascular calcification. Cardiovasc Res. 2011, 91, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeap, B.B.; Chubb, S.A.P.; A McCaul, K.; Flicker, L.; Ho, K.K.Y.; Golledge, J.; Hankey, G.J.; E Norman, P. Associations of IGF1 and its binding proteins with abdominal aortic aneurysm and aortic diameter in older men. Eur. J. Endocrinol. 2012, 166, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Mozo, P.; Rodriguez, C.; Pastor-Vargas, C.; Blanco-Colio, L.; Martinez-Gonzalez, J.; Meilhac, O.; Michel, J.-B.; de Ceniga, M.V.; Egido, J.; Martin-Ventura, J. Plasma profiling by a protein array approach identifies IGFBP-1 as a novel biomarker of abdominal aortic aneurysm. Atherosclerosis 2012, 221, 544–550. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.; Safi, H.; Brasier, A.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef] [Green Version]
- Estrada, A.C.; Irons, L.; Rego, B.V.; Li, G.; Tellides, G.; Humphrey, J.D. Roles of mTOR in thoracic aortopathy understood by complex intracellular signaling interactions. PLOS Comput. Biol. 2021, 17, e1009683. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Zhang, L.; Ren, P.; Nguyen, M.T.; Zou, S.; Wu, D.; Wang, X.L.; Coselli, J.S.; LeMaire, S.A. AKT2 Confers Protection Against Aortic Aneurysms and Dissections. Circ. Res. 2013, 112, 618–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gäbel, G.; Northoff, B.H.; Balboa, A.; Agic, M.B.; Petri, M.; Busch, A.; Maegdefessel, L.; Mahlmann, A.; Ludwig, S.; Teupser, D.; et al. Parallel Murine and Human Aortic Wall Genomics Reveals Metabolic Reprogramming as Key Driver of Abdominal Aortic Aneurysm Progression. J. Am. Hear. Assoc. 2021, 10, e020231. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.; Desdín-Micó, G.; Aranda, J.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.; Roldan-Montero, R.; Acuña, P.; et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Verhagen, J.; Burger, J.; Bekkers, J.; den Dekker, A.; von der Thüsen, J.; Zajec, M.; Brüggenwirth, H.; van der Sterre, M.; van den Born, M.; Luider, T.; et al. Multi-Omics Profiling in Marfan Syndrome: Further Insights into the Molecular Mechanisms Involved in Aortic Disease. Int. J. Mol. Sci. 2021, 23, 438. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Description | Count | p-Value |
|---|---|---|---|---|
| BP | GO:0030335 | positive regulation of cell migration | 3 | 3.56 × 10−4 |
| GO:0048008 | platelet-derived growth factor receptor signaling pathway | 2 | 7.13 × 10−3 | |
| GO:0038083 | peptidyl-tyrosine autophosphorylation | 2 | 1.39 × 10−2 | |
| GO:0008286 | insulin receptor signaling pathway | 2 | 1.39 × 10−2 | |
| GO:0007169 | transmembrane receptor protein tyrosine kinase signaling pathway | 2 | 1.71 × 10−2 | |
| GO:0048015 | phosphatidylinositol-mediated signaling | 2 | 1.88 × 10−2 | |
| GO:0046777 | protein autophosphorylation | 2 | 3.04 × 10−2 | |
| CC | GO:0016020 | membrane | 3 | 4.02 × 10−2 |
| MF | GO:0008286 | insulin receptor substrate binding | 2 | 1.95 × 10−3 |
| GO:0043548 | phosphatidylinositol 3-kinase binding | 2 | 3.37 × 10−3 | |
| GO:0004713 | protein tyrosine kinase activity | 2 | 2.35 × 10−2 |
| Term | Description | Count | p-Value | Genes |
|---|---|---|---|---|
| hsa04014 | Ras signaling pathway | 3 | 9.73 × 10−5 | CSF1, GAB1, IGF1R |
| hsa04151 | PI3K-Akt signaling pathway | 3 | 3.43 × 10−4 | CSF1, JAK2, IGF1R |
| hsa04550 | Signaling pathways regulating pluripotency of stem cells | 2 | 1.82 × 10−3 | JAK2, IGF1R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, X.; Hong, X.; Hong, S.; Huang, Y.; Chen, G.; Chen, Y.; Lin, Y.; Lu, W.; Fu, W.; Wang, L. Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis. Biomedicines 2023, 11, 571. https://doi.org/10.3390/biomedicines11020571
Xie X, Hong X, Hong S, Huang Y, Chen G, Chen Y, Lin Y, Lu W, Fu W, Wang L. Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis. Biomedicines. 2023; 11(2):571. https://doi.org/10.3390/biomedicines11020571
Chicago/Turabian StyleXie, Xinsheng, Xiang Hong, Shichai Hong, Yulong Huang, Gang Chen, Yihui Chen, Yue Lin, Weifeng Lu, Weiguo Fu, and Lixin Wang. 2023. "Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis" Biomedicines 11, no. 2: 571. https://doi.org/10.3390/biomedicines11020571
APA StyleXie, X., Hong, X., Hong, S., Huang, Y., Chen, G., Chen, Y., Lin, Y., Lu, W., Fu, W., & Wang, L. (2023). Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis. Biomedicines, 11(2), 571. https://doi.org/10.3390/biomedicines11020571