

Immune Recognition versus Immune Evasion Systems in Zika Virus Infection
 ,
,  , and
, and
Abstract
:1. Introduction
2. ZIKV Properties
2.1. Gene and Structure
2.2. African and Asian ZIKV Lineages
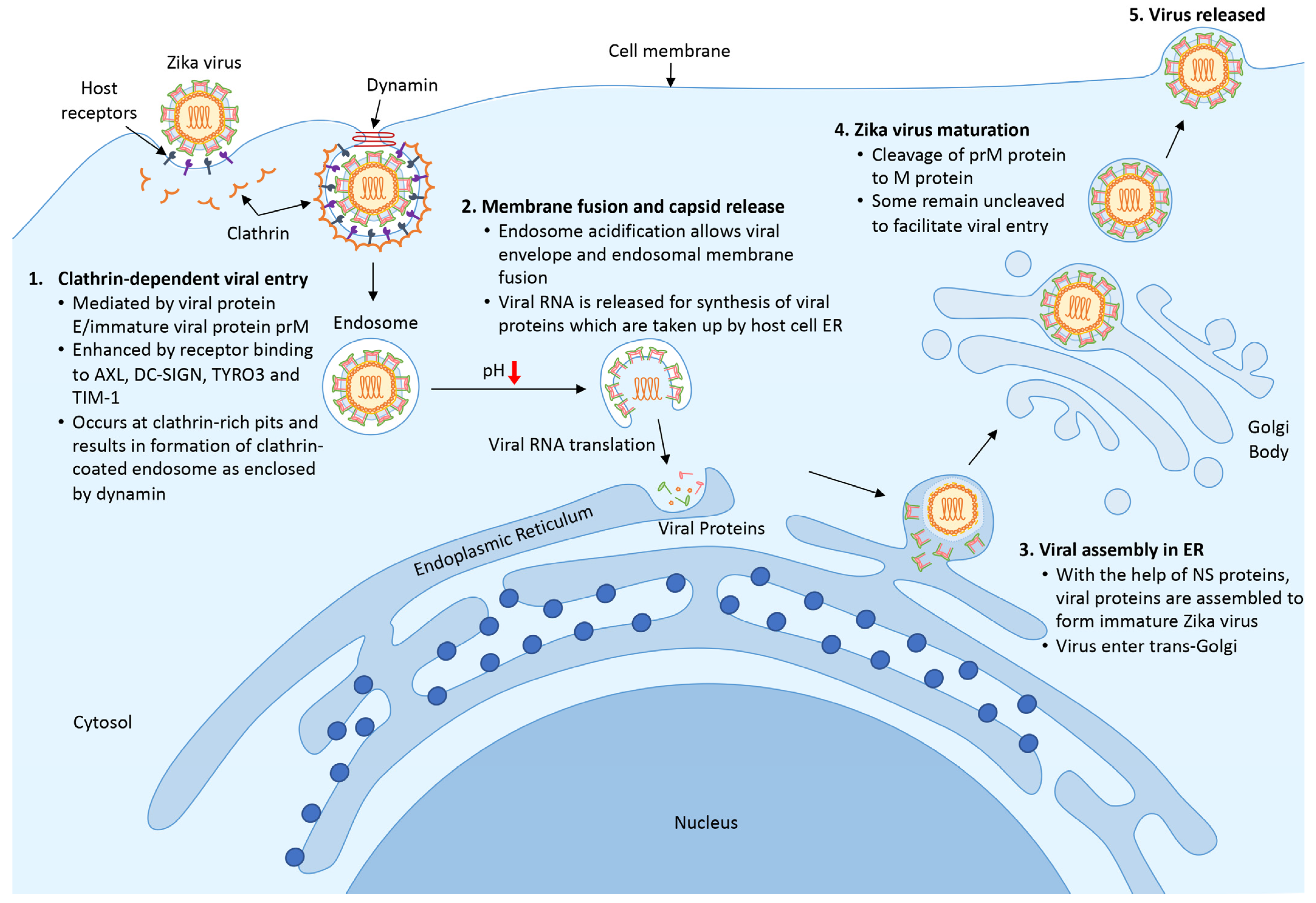
2.3. Transmission and Life Cycle
2.4. Symptoms Caused by ZIKV Infection
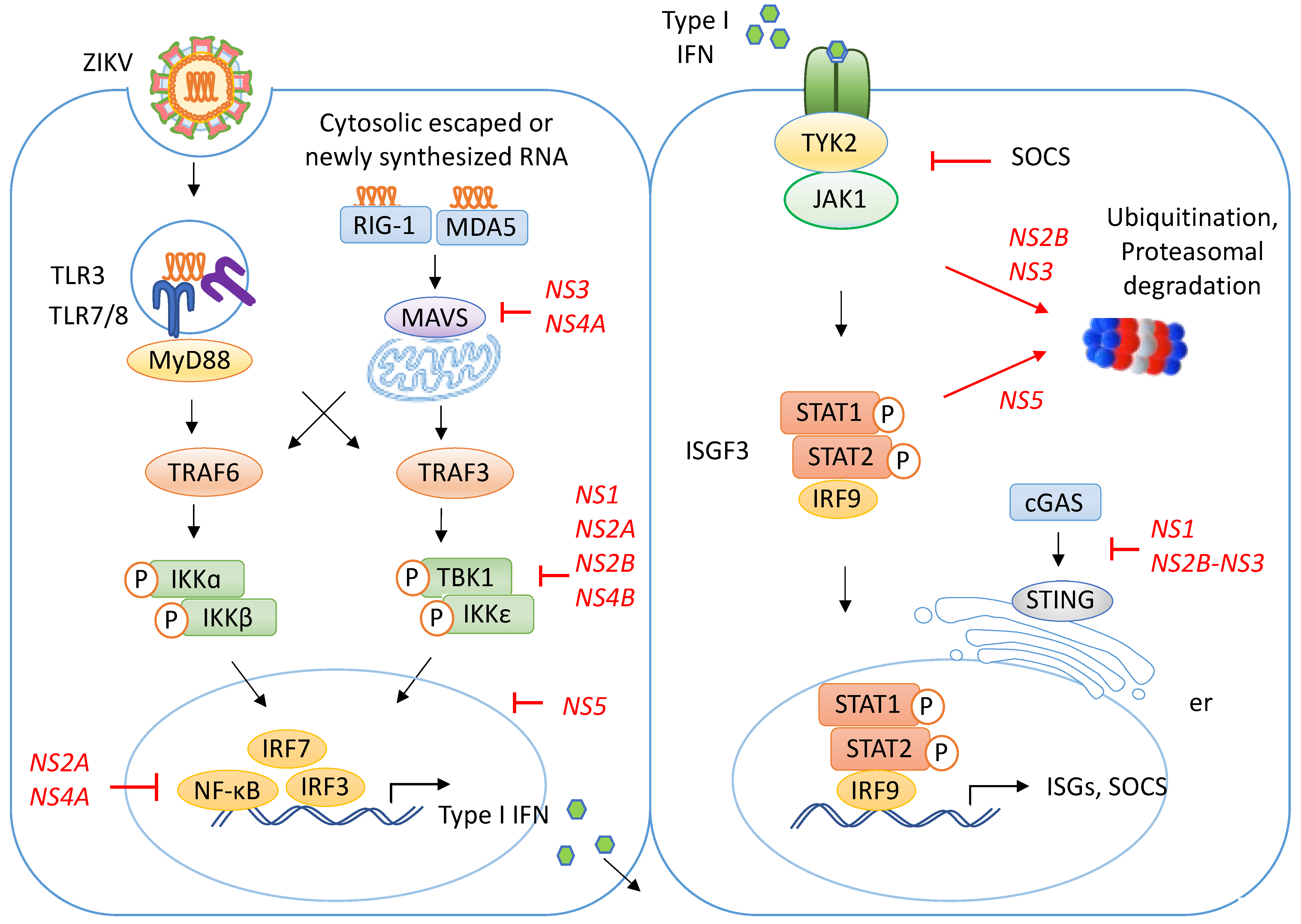
3. Innate Immune Recognition
3.1. ZIKV Recognition by RIG-I Receptor
3.2. ZIKV Recognition by TLR3
3.3. ZIKV Recognition by TLR7/8
3.4. Signaling Pathway Activated by ZIKV Recognition
3.5. Low Pattern-Recognition Receptors in the Lower Female Reproductive Tract Enables Viral Replication
4. ZIKV Attenuates Innate Recognition
4.1. ZIKV Modulates the Translocation of RIG-I and MDA5
4.2. ZIKV Degrades the cGAS/STING Pathway
4.3. ZIKV Blocks TBK1 Phosphorylation
4.4. ZIKV Represses the Promoter Activity of the NF-κB and IRF-3 Transcription Factors
4.5. ZIKV Disrupts the JAK/STAT Signaling Pathway
4.6. ZIKV Suppresses Type I IFN Signaling through Inducing Inflammasome Activity
4.7. ZIKV Antagonizes RNAi-Mediated Antiviral Activity
5. Other Evasion Strategies Exploited by ZIKV
5.1. ZIKV Evades Immune Attack through Gene Mutation
5.2. ZIKV Alters Cellular Processes
5.3. ZIKV Forms an RNA Cap through Methyltransferase Activity
5.4. ZIKV-Mediated Modulation of Humoral Immune Response
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Smithburn, K.C. Neutralizing antibodies against certain recently isolated viruses in the sera of human beings residing in East Africa. J. Immunol. 1952, 69, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and Emergence of Zika Virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef] [Green Version]
- WHO. Situation Report. Zika Virus Microcephaly Guillain-Barré Syndorme; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Lum, F.-M.; Low, D.K.S.; Fan, Y.; Tan, J.J.L.; Lee, B.; Chan, J.K.Y.; Rénia, L.; Ginhoux, F.; Ng, L.F.P. Zika Virus Infects Human Fetal Brain Microglia and Induces Inflammation. Clin. Infect. Dis. 2017, 64, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-C.; Abraham, R.; Shim, B.-S.; Choe, H.; Page, D.T. Zika virus infection during the period of maximal brain growth causes microcephaly and corticospinal neuron apoptosis in wild type mice. Sci. Rep. 2016, 6, 34793. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolic, M.; et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell Stem Cell 2017, 20, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontera, J.A.; da Silva, I.R. Zika Getting on Your Nerves? The Association with the Guillain-Barre Syndrome. N. Engl. J. Med. 2016, 375, 1581–1582. [Google Scholar] [CrossRef] [Green Version]
- Yung, C.F.; Thoon, K.C. Guillain-Barre Syndrome and Zika Virus: Estimating Attributable Risk to Inform Intensive Care Capacity Preparedness. Clin. Infect. Dis. 2016, 63, 708–709. [Google Scholar] [CrossRef] [Green Version]
- Brasil, P.; Sequeira, P.C.; Freitas, A.D.; Zogbi, H.E.; Calvet, G.A.; de Souza, R.V.; Siqueira, A.M.; de Mendonca, M.C.; Nogueira, R.M.; de Filippis, A.M.; et al. Guillain-Barre syndrome associated with Zika virus infection. Lancet 2016, 387, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, J. Study links Zika virus to Guillain-Barre syndrome. BMJ 2016, 352, i1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.F.; Xu, Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.A.; Mier-y-Teran-Romero, L.; Reefhuis, J.; Gilboa, S.M.; Hills, S.L. Zika and the Risk of Microcephaly. N. Engl. J. Med. 2016, 375, 1–4. [Google Scholar] [CrossRef]
- Rodrigues, L.C. Microcephaly and Zika virus infection. Lancet 2016, 387, 2070–2072. [Google Scholar] [CrossRef] [Green Version]
- Rubin, E.J.; Greene, M.F.; Baden, L.R. Zika Virus and Microcephaly. N. Engl. J. Med. 2016, 374, 984–985. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Wang, Y.; Ling, L.; Zhang, Z.; Marin-Lopez, A. Current Advances in Zika Vaccine Development. Vaccines 2022, 10, 1816. [Google Scholar] [CrossRef]
- Cheong, H.C.; Cheok, Y.Y.; Chan, Y.T.; Sulaiman, S.; Looi, C.Y.; Alshanon, A.F.; Hassan, J.; Abubakar, S.; Wong, W.F. Zika Virus Vaccine: The Current State of Affairs and Challenges Posed by Antibody-Dependent Enhancement Reaction. Viral. Immunol. 2022, 35, 586–596. [Google Scholar] [CrossRef]
- Wang, A.; Thurmond, S.; Islas, L.; Hui, K.; Hai, R. Zika virus genome biology and molecular pathogenesis. Emerg. Microbes Infect. 2017, 6, e13. [Google Scholar] [CrossRef] [Green Version]
- Enfissi, A.; Codrington, J.; Roosblad, J.; Kazanji, M.; Rousset, D. Zika virus genome from the Americas. Lancet 2016, 387, 227–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-Y.; Shi, W.-F.; Qin, C.-F. The evolution of Zika virus from Asia to the Americas. Nat. Rev. Microbiol. 2019, 17, 131–139. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, S.; Li, T.; McCune, B.T.; Edeling, M.A.; Fremont, D.H.; Cristea, I.M.; Diamond, M.S. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 2012, 86, 7360–7371. [Google Scholar] [CrossRef] [Green Version]
- Hilgenfeld, R. Zika virus NS1, a pathogenicity factor with many faces. EMBO J. 2016, 35, 2631–2633. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Jurado, S.; Nogales, A.; Avila-Perez, G.; Iborra, F.J.; Martinez-Sobrido, L.; Almazan, F. An Alanine-to-Valine Substitution in the Residue 175 of Zika Virus NS2A Protein Affects Viral RNA Synthesis and Attenuates the Virus In Vivo. Viruses 2018, 10, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xie, X.; Xia, H.; Zou, J.; Huang, L.; Popov, V.L.; Chen, X.; Shi, P.-Y. Zika Virus NS2A-Mediated Virion Assembly. mBio 2019, 10, e02375-02319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xie, X.; Zou, J.; Xia, H.; Shan, C.; Chen, X.; Shi, P.-Y. Genetic and biochemical characterizations of Zika virus NS2A protein. Emerg. Microbes Infect. 2019, 8, 585–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, K.-J.; Song, G.; Qian, X.; Pan, J.; Xu, D.; Rho, H.-S.; Kim, N.-S.; Habela, C.; Zheng, L.; Jacob, F.; et al. Zika-Virus-Encoded NS2A Disrupts Mammalian Cortical Neurogenesis by Degrading Adherens Junction Proteins. Cell Stem Cell 2017, 21, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Sampath, A.; Padmanabhan, R. Molecular targets for flavivirus drug discovery. Antivir. Res. 2009, 81, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Noble, C.G.; Seh, C.C.; Chao, A.T.; Shi, P.Y. Ligand-Bound Structures of the Dengue Virus Protease Reveal the Active Conformation. J. Virol. 2012, 86, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef] [Green Version]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced Autophagy Protects Cells against Death and Enhances Virus Replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 2014, 8, e2636. [Google Scholar] [CrossRef] [Green Version]
- Haddow, A.D.; Nasar, F.; Guzman, H.; Ponlawat, A.; Jarman, R.G.; Tesh, R.B.; Weaver, S.C. Genetic Characterization of Spondweni and Zika Viruses and Susceptibility of Geographically Distinct Strains of Aedes aegypti, Aedes albopictus and Culex quinquefasciatus (Diptera: Culicidae) to Spondweni Virus. PLoS Negl. Trop. Dis. 2016, 10, e0005083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvez, E.; O'Connor, O.; Pol, M.; Rousset, D.; Faye, O.; Richard, V.; Tarantola, A.; Dupont-Rouzeyrol, M. Differential transmission of Asian and African Zika virus lineages by Aedes aegypti from New Caledonia. Emerg. Microbes. Infect. 2018, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Yin, J. Kinetics of Asian and African Zika virus lineages over single-cycle and multi-cycle growth in culture: Gene expression, cell killing, virus production, and mathematical modeling. Biotechnol. Bioeng. 2021, 118, 4231–4245. [Google Scholar] [CrossRef]
- Simonin, Y.; van Riel, D.; Van de Perre, P.; Rockx, B.; Salinas, S. Differential virulence between Asian and African lineages of Zika virus. PLoS Negl. Trop. Dis. 2017, 11, e0005821. [Google Scholar] [CrossRef]
- Aubry, F.; Jacobs, S.; Darmuzey, M.; Lequime, S.; Delang, L.; Fontaine, A.; Jupatanakul, N.; Miot, E.F.; Dabo, S.; Manet, C.; et al. Recent African strains of Zika virus display higher transmissibility and fetal pathogenicity than Asian strains. Nat. Commun. 2021, 12, 916. [Google Scholar] [CrossRef]
- Sheridan, M.A.; Balaraman, V.; Schust, D.J.; Ezashi, T.; Roberts, R.M.; Franz, A.W.E. African and Asian strains of Zika virus differ in their ability to infect and lyse primitive human placental trophoblast. PLoS ONE 2018, 13, e0200086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic Differences between Asian and African Lineage Zika Viruses in Human Neural Progenitor Cells. mSphere 2017, 2, e00292-17. [Google Scholar] [CrossRef] [Green Version]
- WHO. Zika Epidemiology Update; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Shan, C.; Xia, H.; Haller, S.L.; Azar, S.R.; Liu, Y.; Liu, J.; Muruato, A.E.; Chen, R.; Rossi, S.L.; Wakamiya, M.; et al. A Zika virus envelope mutation preceding the 2015 epidemic enhances virulence and fitness for transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 20190–20197. [Google Scholar] [CrossRef]
- Khan, E.; Jindal, H.; Mishra, P.; Suvvari, T.K.; Jonna, S. The 2021 Zika outbreak in Uttar Pradesh state of India: Tackling the emerging public health threat. Trop. Doct. 2022, 52, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, M.; Pramanik, D.; Riyaz, R.; Hasan, M.M.; Essar, M.Y. Dual burden of Zika and COVID-19 in India: Challenges, opportunities and recommendations. Trop. Med. Health 2021, 49, 83. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Malhotra, B.; Sapkal, G.; Nyayanit, D.A.; Deshpande, G.; Gupta, N.; Padinjaremattathil, U.T.; Sharma, H.; Sahay, R.R.; Sharma, P.; et al. Zika virus outbreak in Rajasthan, India in 2018 was caused by a virus endemic to Asia. Infect. Genet. Evol. 2019, 69, 199–202. [Google Scholar] [CrossRef]
- Thangamani, S.; Huang, J.; Hart, C.E.; Guzman, H.; Tesh, R.B. Vertical Transmission of Zika Virus in Aedes aegypti Mosquitoes. Am. J. Trop. Med. Hyg. 2016, 95, 1169–1173. [Google Scholar] [CrossRef] [Green Version]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. = Eur. Commun. Dis. Bull. 2014, 19, 20761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, J.; Peixoto, T.M.; Siqueira, A.M.; Lamas, C.C. Sexually acquired Zika virus: A systematic review. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2017, 23, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Prisant, N.; Bujan, L.; Benichou, H.; Hayot, P.-H.; Pavili, L.; Lurel, S.; Herrmann, C.; Janky, E.; Joguet, G. Zika virus in the female genital tract. Lancet Infect. Dis. 2016, 16, 1000–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciota, A.T.; Bialosuknia, S.M.; Ehrbar, D.J.; Kramer, L.D. Vertical Transmission of Zika Virus by Aedes aegypti and Ae. albopictus Mosquitoes. Emerg. Infect. Dis. 2017, 23, 880–882. [Google Scholar] [CrossRef]
- Persaud, M.; Martinez-Lopez, A.; Buffone, C.; Porcelli, S.A.; Diaz-Griffero, F. Infection by Zika viruses requires the transmembrane protein AXL, endocytosis and low pH. Virology 2018, 518, 301–312. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrelli, A.; de Moura, R.R.; Crovella, S.; Brandao, L.A.C. ZIKA virus entry mechanisms in human cells. Infect. Genet. Evol. 2019, 69, 22–29. [Google Scholar] [CrossRef]
- Rey, F.A.; Stiasny, K.; Heinz, F.X. Flavivirus structural heterogeneity: Implications for cell entry. Curr. Opin. Virol. 2017, 24, 132–139. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika Virus Outbreak on Yap Island, Federated States of Micronesia. N. Eng. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, W.K.; Carmo, E.H.; Henriques, C.M.; Coelho, G.; Vazquez, E.; Cortez-Escalante, J.; Molina, J.; Aldighieri, S.; Espinal, M.A.; Dye, C. Zika Virus Infection and Associated Neurologic Disorders in Brazil. N. Engl. J. Med. 2017, 376, 1591–1593. [Google Scholar] [CrossRef] [PubMed]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef] [PubMed]
- Schwartzmann, P.V.; Ramalho, L.N.Z.; Neder, L.; Vilar, F.C.; Ayub-Ferreira, S.M.; Romeiro, M.F.; Takayanagui, O.M.; dos Santos, A.C.; Schmidt, A.; Figueiredo, L.T.M.; et al. Zika Virus Meningoencephalitis in an Immunocompromised Patient. Mayo Clin. Proc. 2017, 92, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mécharles, S.; Herrmann, C.; Poullain, P.; Tran, T.-H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A.J.T.L. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef] [Green Version]
- Yepez, J.B.; Murati, F.A.; Pettito, M.; Peñaranda, C.F.; de Yepez, J.; Maestre, G.; Arevalo, J.F.; for The Johns Hopkins Zika, C. Ophthalmic Manifestations of Congenital Zika Syndrome in Colombia and Venezuela. JAMA Ophthalmol. 2017, 135, 440–445. [Google Scholar] [CrossRef]
- Tsui, I.; Moreira, M.E.L.; Rossetto, J.D.; Vasconcelos, Z.; Gaw, S.L.; Neves, L.M.; Zin, O.A.; Haefeli, L.; Silveira Filho, J.C.B.; Gomes, S.C.; et al. Eye Findings in Infants with Suspected or Confirmed Antenatal Zika Virus Exposure. Pediatrics 2018, 142, e20181104. [Google Scholar] [CrossRef] [Green Version]
- Lopes Moreira, M.E.; Nielsen-Saines, K.; Brasil, P.; Kerin, T.; Damasceno, L.; Pone, M.; Carvalho, L.M.A.; Pone, S.M.; Vasconcelos, Z.; Ribeiro, I.P.; et al. Neurodevelopment in Infants Exposed to Zika Virus in Utero. N. Engl. J. Med. 2018, 379, 2377–2379. [Google Scholar] [CrossRef]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Raper, J.; Kovacs-Balint, Z.; Mavigner, M.; Gumber, S.; Burke, M.W.; Habib, J.; Mattingly, C.; Fair, D.; Earl, E.; Feczko, E. Long-term alterations in brain and behavior after postnatal Zika virus infection in infant macaques. Nat. Commun. 2020, 11, 2534. [Google Scholar] [CrossRef]
- Romani, N.; Pieras, M.; Frick, M.A.; Sulleiro, E.; Rodo, C.; Silgado, A.; Suy, A.; Espiau, M.; Thorne, C.; Giaquinto, C.; et al. Neurological Short-Term Outcomes of a Cohort of Children Born to Zika Virus-Infected Mothers in Barcelona. Children 2022, 9, 537. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.T.M.; Hamanaka, T.; Pone, S.; Aibe, M.S.; Gomes, S.C.; Nielsen-Saines, K.; Brickley, E.B.; Moreira, M.E.; Pone, M. Gross motor function in children with Congenital Zika Syndrome from Rio de Janeiro, Brazil. Eur. J. Pediatr. 2022, 181, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell'Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, C.; McKernan, D.P. Anti-Viral Pattern Recognition Receptors as Therapeutic Targets. Cells 2021, 10, 2258. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Chang, T.H.; Liao, C.L.; Lin, Y.L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Plociennikowska, A.; Frankish, J.; Moraes, T.; Del Prete, D.; Kahnt, F.; Acuna, C.; Slezak, M.; Binder, M.; Bartenschlager, R. TLR3 activation by Zika virus stimulates inflammatory cytokine production which dampens the antiviral response induced by RIG-I-like receptors. J. Virol. 2021, 95, e01050-20. [Google Scholar] [CrossRef]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Loriere, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5' Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M., Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Berke, I.C.; Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012, 31, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- da Silva, M.H.M.; Moises, R.N.C.; Alves, B.E.B.; Pereira, H.W.B.; de Paiva, A.A.P.; Morais, I.C.; Nascimento, Y.M.; Monteiro, J.D.; de Souto, J.T.; Nascimento, M.S.L.; et al. Innate immune response in patients with acute Zika virus infection. Med. Microbiol. Immunol. 2019, 208, 703–714. [Google Scholar] [CrossRef]
- Ojha, C.R.; Rodriguez, M.; Karuppan, M.K.M.; Lapierre, J.; Kashanchi, F.; El-Hage, N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PLoS ONE 2019, 14, e0208543. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.N.O.; Ribeiro, D.R.; Cardoso Alves, J.; Cazzaniga, R.A.; Magalhaes, L.S.; de Souza, M.S.F.; Fonseca, A.B.L.; Bispo, A.J.B.; Porto, R.L.S.; Santos, C.A.D.; et al. Association Between Zika Virus Microcephaly in Newborns With the rs3775291 Variant in Toll-Like Receptor 3 and rs1799964 Variant at Tumor Necrosis Factor-alpha Gene. J. Infect. Dis. 2019, 220, 1797–1801. [Google Scholar] [CrossRef]
- Zhou, P.; Fan, L.; Yu, K.D.; Zhao, M.W.; Li, X.X. Toll-like receptor 3 C1234T may protect against geographic atrophy through decreased dsRNA binding capacity. FASEB J. 2011, 25, 3489–3495. [Google Scholar] [CrossRef]
- Vanwalscappel, B.; Tada, T.; Landau, N.R. Toll-like receptor agonist R848 blocks Zika virus replication by inducing the antiviral protein viperin. Virology 2018, 522, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Sharma, S.; Grandvaux, N.; Zhou, G.-P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raftery, N.; Stevenson, N.J. Advances in anti-viral immune defence: Revealing the importance of the IFN JAK/STAT pathway. Cell Mol. Life Sci. 2017, 74, 2525–2535. [Google Scholar] [CrossRef]
- Meylan, E.; Tschopp, J.; Karin, M. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Gao, C.; Wen, C.; Zou, P.; Qi, X.; Cardona, C.J.; Xing, Z. Intrinsic features of Zika Virus non-structural proteins NS2A and NS4A in the regulation of viral replication. PLoS Negl. Trop. Dis. 2022, 16, e0010366. [Google Scholar] [CrossRef]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Winkler, C.W.; Clancy, C.S.; Rosenke, R.; Peterson, K.E. Zika virus vertical transmission in interferon receptor1-antagonized Rag1(-/-) mice results in postnatal brain abnormalities and clinical disease. Acta Neuropathol. Commun. 2022, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Lew, I.; Wu, F.; Fritts, L.; Fontaine, K.A.; Tomar, S.; Trapecar, M.; Shehata, H.M.; Ott, M.; Miller, C.J.; et al. Low expression of RNA sensors impacts Zika virus infection in the lower female reproductive tract. Nat. Commun. 2019, 10, 4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Dong, X.; He, Z.; Wu, Y.; Zhang, S.; Lin, J.; Yang, Y.; Chen, J.; An, S.; Yin, Y.; et al. Zika virus antagonizes interferon response in patients and disrupts RIG-I-MAVS interaction through its CARD-TM domains. Cell Biosci. 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; PLoSs, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Nguyen, T.T.N.; Myoung, J. Zika Virus-Encoded NS2A and NS4A Strongly Downregulate NF-kappaB Promoter Activity. J. Microbiol. Biotechnol. 2020, 30, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Gavegnano, C.; Bassit, L.C.; Cox, B.D.; Hsiao, H.M.; Johnson, E.L.; Suthar, M.; Chakraborty, R.; Schinazi, R.F. Jak Inhibitors Modulate Production of Replication-Competent Zika Virus in Human Hofbauer, Trophoblasts, and Neuroblastoma cells. Pathog. Immun. 2017, 2, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Fanunza, E.; Carletti, F.; Quartu, M.; Grandi, N.; Ermellino, L.; Milia, J.; Corona, A.; Capobianchi, M.R.; Ippolito, G.; Tramontano, E. Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence 2021, 12, 1580–1596. [Google Scholar] [CrossRef] [PubMed]
- Fanunza, E.; Grandi, N.; Quartu, M.; Carletti, F.; Ermellino, L.; Milia, J.; Corona, A.; Capobianchi, M.R.; Ippolito, G.; Tramontano, E. INMI1 Zika Virus NS4B Antagonizes the Interferon Signaling by Suppressing STAT1 Phosphorylation. Viruses 2021, 13, 2448. [Google Scholar] [CrossRef]
- Slonchak, A.; Wang, X.; Aguado, J.; Sng, J.D.J.; Chaggar, H.; Freney, M.E.; Yan, K.; Torres, F.J.; Amarilla, A.A.; Balea, R.; et al. Zika virus noncoding RNA cooperates with the viral protein NS5 to inhibit STAT1 phosphorylation and facilitate viral pathogenesis. Sci. Adv. 2022, 8, eadd8095. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, S.; Balasubramaniam, V.R.; Brown, J.A.; Mena, I.; Grant, A.; Bardina, S.V.; Maringer, K.; Schwarz, M.C.; Maestre, A.M.; Sourisseau, M.; et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 2017, 13, e1006258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yang, Y.-f.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.-l.; Cui, Y.-r.; Bai, R.; Liang, Y.-j. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef]
- He, Z.; Chen, J.; Zhu, X.; An, S.; Dong, X.; Yu, J.; Zhang, S.; Wu, Y.; Li, G.; Zhang, Y.; et al. NLRP3 Inflammasome Activation Mediates Zika Virus-Associated Inflammation. J. Infect. Dis. 2018, 217, 1942–1951. [Google Scholar] [CrossRef]
- Wang, W.; Li, G.; De, W.; Luo, Z.; Pan, P.; Tian, M.; Wang, Y.; Xiao, F.; Li, A.; Wu, K.; et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat. Commun. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Sci. 2013, 342, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, Y.; Ding, S.W. Small RNA-based antimicrobial immunity. Nat. Rev. Immunol. 2019, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zeng, J. Neuroimmune Evasion of Zika Virus to Facilitate Viral Pathogenesis. Front Cell Infect. Microbiol. 2021, 11, 662447. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, Y.P.; Wang, M.; Miao, M.; Zhou, H.; Xu, J.; Kong, J.; Zheng, D.; Li, R.T.; Zhang, R.R.; et al. Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci. Adv. 2020, 6, eaax7989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Dong, S.; Luo, Z.; Xie, X.; Fu, B.; Li, P.; Liu, C.; Yang, X.; Chen, Y.; Wang, X.; et al. The Zika Virus Capsid Disrupts Corticogenesis by Suppressing Dicer Activity and miRNA Biogenesis. Cell Stem Cell 2020, 27, 618–632. [Google Scholar] [CrossRef]
- Zeng, J.; Luo, Z.; Dong, S.; Xie, X.; Liang, X.; Yan, Y.; Liang, Q.; Zhao, Z. Functional Mapping of AGO-Associated Zika Virus-Derived Small Interfering RNAs in Neural Stem Cells. Front. Cell Infect. Microbiol. 2021, 11, 628887. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.P.; Qiu, Y.; Zhang, B.; Chen, G.; Chen, Q.; Wang, M.; Mo, F.; Xu, J.; Wu, J.; Zhang, R.R.; et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res. 2019, 29, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Dang, J.W.; Tiwari, S.K.; Qin, Y.; Rana, T.M. Genome-wide Integrative Analysis of Zika-Virus-Infected Neuronal Stem Cells Reveals Roles for MicroRNAs in Cell Cycle and Stemness. Cell Rep. 2019, 27, 3618–3628. [Google Scholar] [CrossRef] [Green Version]
- Kozak, R.A.; Majer, A.; Biondi, M.J.; Medina, S.J.; Goneau, L.W.; Sajesh, B.V.; Slota, J.A.; Zubach, V.; Severini, A.; Safronetz, D.; et al. MicroRNA and mRNA Dysregulation in Astrocytes Infected with Zika Virus. Viruses 2017, 9, 297. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Huang, X.-Y.; Liu, Z.-Y.; Zhang, F.; Zhu, X.-L.; Yu, J.-Y.; Ji, X.; Xu, Y.-P.; Li, G.; Li, C. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regla-Nava, J.A.; Wang, Y.T.; Fontes-Garfias, C.R.; Liu, Y.; Syed, T.; Susantono, M.; Gonzalez, A.; Viramontes, K.M.; Verma, S.K.; Kim, K.; et al. A Zika virus mutation enhances transmission potential and confers escape from protective dengue virus immunity. Cell Rep. 2022, 39, 110655. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.F.; Zhang, R.; Wang, T.; Qin, C.F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482–486. [Google Scholar] [CrossRef] [Green Version]
- May, M.; Relich, R.F. A comprehensive systems biology approach to studying Zika virus. PloS ONE 2016, 11, e0161355. [Google Scholar] [CrossRef] [Green Version]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Gorshkov, K.; Lee, E.M.; Xu, M.; Cheng, Y.S.; Sun, N.; Soheilian, F.; de Val, N.; Ming, G.; Song, H.; et al. Zika Virus-Induced Neuronal Apoptosis via Increased Mitochondrial Fragmentation. Front. Microbiol. 2020, 11, 598203. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Conde, J.N.; da Silva, E.M.; Allonso, D.; Coelho, D.R.; da Silva Andrade, I.; de Medeiros, L.N.; Menezes, J.L.; Barbosa, A.S.; Mohana-Borges, R. Inhibition of the membrane attack complex by dengue virus NS1 through interaction with vitronectin and terminal complement proteins. J. Virol. 2016, 90, 9570–9581. [Google Scholar] [CrossRef] [Green Version]
- Glasner, A.; Oiknine-Djian, E.; Weisblum, Y.; Diab, M.; Panet, A.; Wolf, D.G.; Mandelboim, O. Zika virus escapes NK cell detection by upregulating major histocompatibility complex class I molecules. J. Virol. 2017, 91, e00785-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef] [Green Version]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Chang, D.C.; Hoang, L.T.; Naim, A.N.M.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.-Y. Evasion of early innate immune response by 2′-O-methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef]
- Coutard, B.; Barral, K.; Lichière, J.; Selisko, B.; Martin, B.; Aouadi, W.; Lombardia, M.O.; Debart, F.; Vasseur, J.-J.; Guillemot, J.C. Zika virus methyltransferase: Structure and functions for drug design perspectives. J. Virol. 2017, 91, e02202-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Acosta, R.; Xie, X.; Shan, C.; Baker, C.K.; Shi, P.-Y.; Rossi, S.L.; Garcia-Blanco, M.A.; Bradrick, S. Fragile X mental retardation protein is a Zika virus restriction factor that is antagonized by subgenomic flaviviral RNA. Elife 2018, 7, e39023. [Google Scholar] [CrossRef]
- Almansour, I.; Alfares, R.; Aljofi, H. Large-scale analysis of B-cell epitopes of envelope: Implications for Zika vaccine and immunotherapeutic development. F1000Research 2018, 7, 1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, B.O.; Sachs, D.; Schwarz, M.C.; Palese, P.; Evans, M.J. Transposon Mutagenesis of the Zika Virus Genome Highlights Regions Essential for RNA Replication and Restricted for Immune Evasion. J. Virol. 2017, 91, e00698-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Host Immune Pathways | ZIKV Protein | Mechanism | References |
|---|---|---|---|
| RIG-I and MDA5 signaling | NS4A |
| [108] |
| NS3 |
| [109] | |
| cGAS/STING pathway | NS1 |
| [107] |
| NS2B-NS3 |
| [112] | |
| TBK1 phosphorylation | NS1 |
| [86,112] |
| NS2A, NS2B, and NS4B |
| [112] | |
| NS4A |
| [112] | |
| NF-κB and IRF-3 | NS2A and NS4A |
| [113] |
| NS5 |
| [112] | |
| JAK/STAT pathway | NS2B-NS3 |
| [114] |
| NS5 |
| [119,120] | |
| NS2A |
| [116] | |
| NS4B |
| [117,118] | |
| NS5-noncoding RNA |
| [117,118] | |
| Type I IFN signaling | NS1 |
| [110] |
| NS5 |
| [123,124] | |
| Antiviral RNAi | NS2A |
| [128] |
| C |
| [129,130] | |
| Gene mutation | pRM |
| [134] |
| NS2B |
| [135] | |
| NS1 |
| [136] | |
| E |
| [137] | |
| Cellular processes | NS4B |
| [140] |
| - |
| [70] [139] | |
| NS4A and NS4B |
| [36] | |
| NS1 |
| [141] | |
| - |
| [142] | |
| Methyltransferase activity | NS5 |
| [146] |
| Humoral immunity | RdRP |
| [148] |
| E |
| [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, Y.T.; Cheok, Y.Y.; Cheong, H.C.; Tang, T.F.; Sulaiman, S.; Hassan, J.; Looi, C.Y.; Tan, K.-K.; AbuBakar, S.; Wong, W.F. Immune Recognition versus Immune Evasion Systems in Zika Virus Infection. Biomedicines 2023, 11, 642. https://doi.org/10.3390/biomedicines11020642
Chan YT, Cheok YY, Cheong HC, Tang TF, Sulaiman S, Hassan J, Looi CY, Tan K-K, AbuBakar S, Wong WF. Immune Recognition versus Immune Evasion Systems in Zika Virus Infection. Biomedicines. 2023; 11(2):642. https://doi.org/10.3390/biomedicines11020642
Chicago/Turabian StyleChan, Yee Teng, Yi Ying Cheok, Heng Choon Cheong, Ting Fang Tang, Sofiah Sulaiman, Jamiyah Hassan, Chung Yeng Looi, Kim-Kee Tan, Sazaly AbuBakar, and Won Fen Wong. 2023. "Immune Recognition versus Immune Evasion Systems in Zika Virus Infection" Biomedicines 11, no. 2: 642. https://doi.org/10.3390/biomedicines11020642
APA StyleChan, Y. T., Cheok, Y. Y., Cheong, H. C., Tang, T. F., Sulaiman, S., Hassan, J., Looi, C. Y., Tan, K. -K., AbuBakar, S., & Wong, W. F. (2023). Immune Recognition versus Immune Evasion Systems in Zika Virus Infection. Biomedicines, 11(2), 642. https://doi.org/10.3390/biomedicines11020642