
Molecular Screening of Bioactive Compounds of Garlic for Therapeutic Effects against COVID-19
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
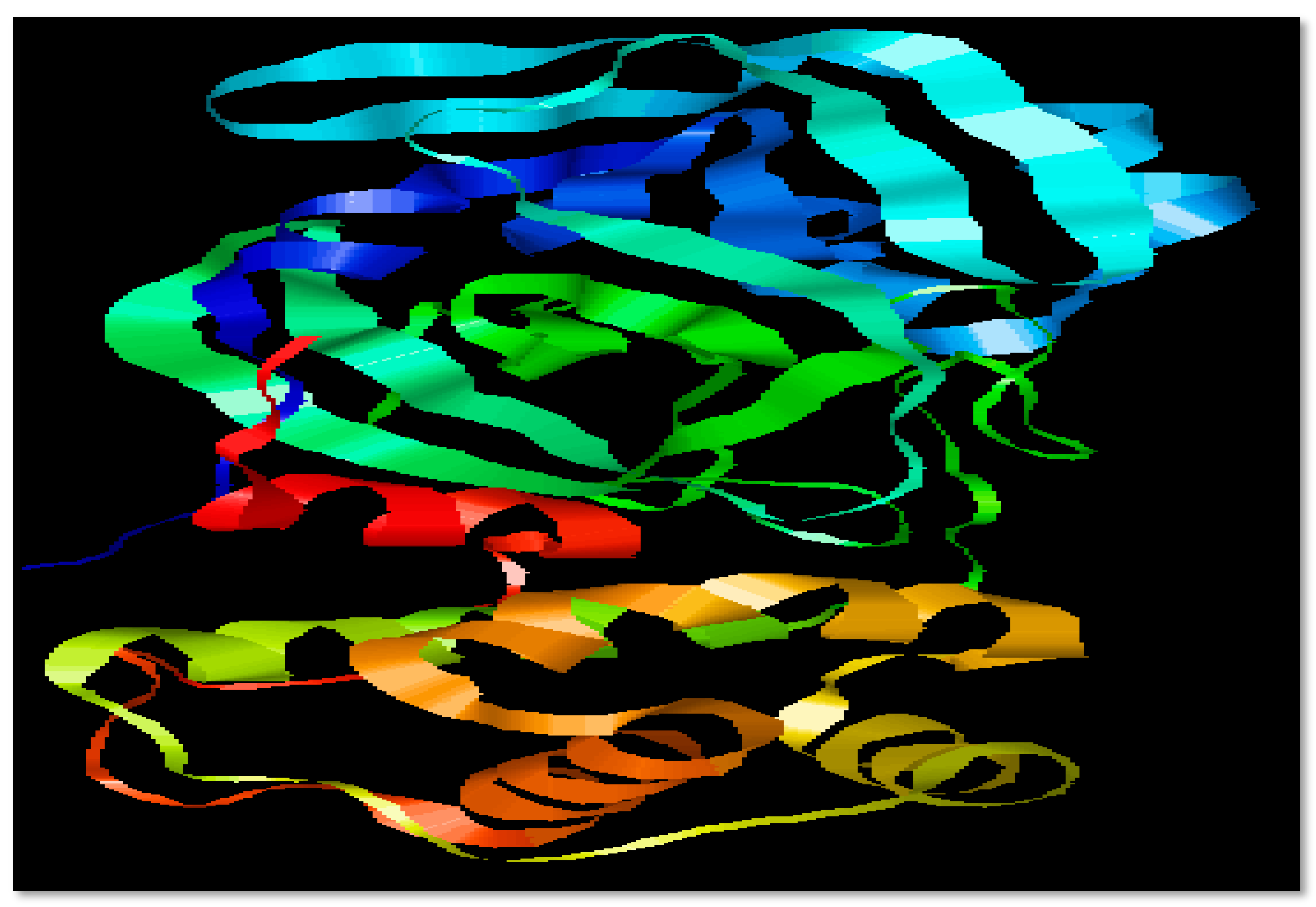
2.1. Selection of Protein
2.2. Primary Sequence Retrieval
2.3. Analysis of Physiochemical Properties
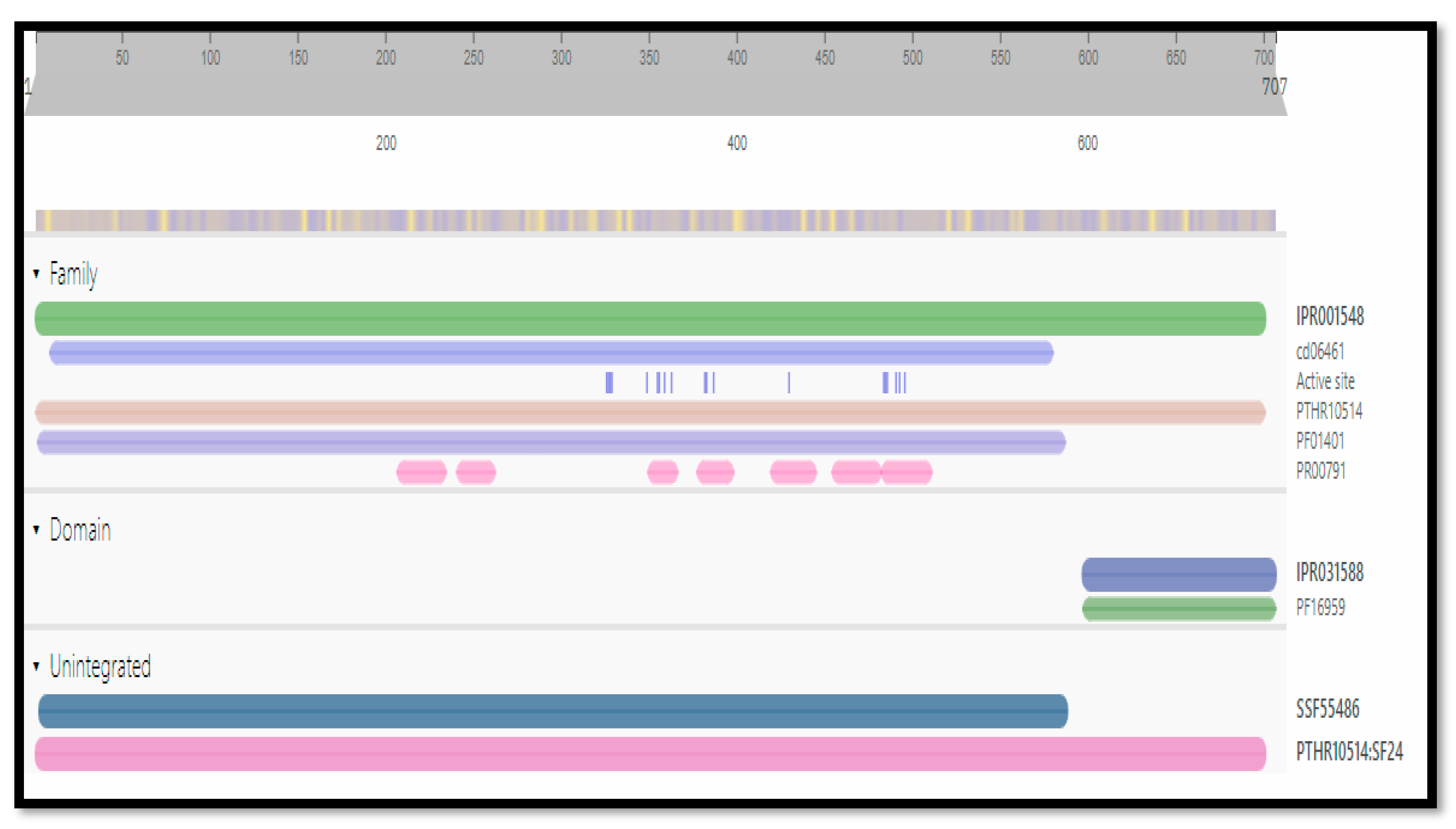
2.4. Identification of Functional Domains
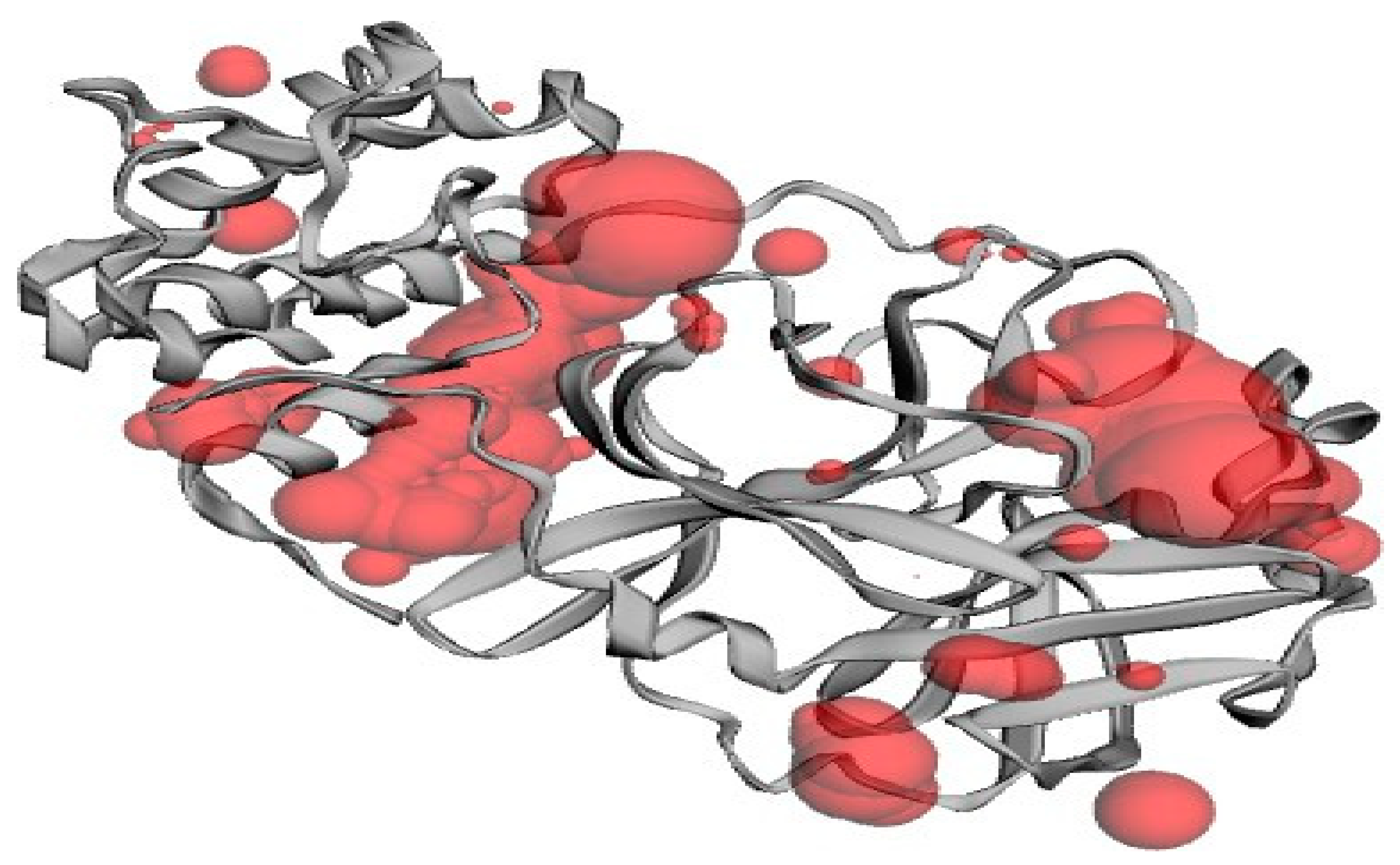
2.5. Active Site Identification
2.6. Ligand Preparation
2.7. Bioactivity Analysis of Ligands and Toxicity Measurement
2.8. Molecular Docking Process
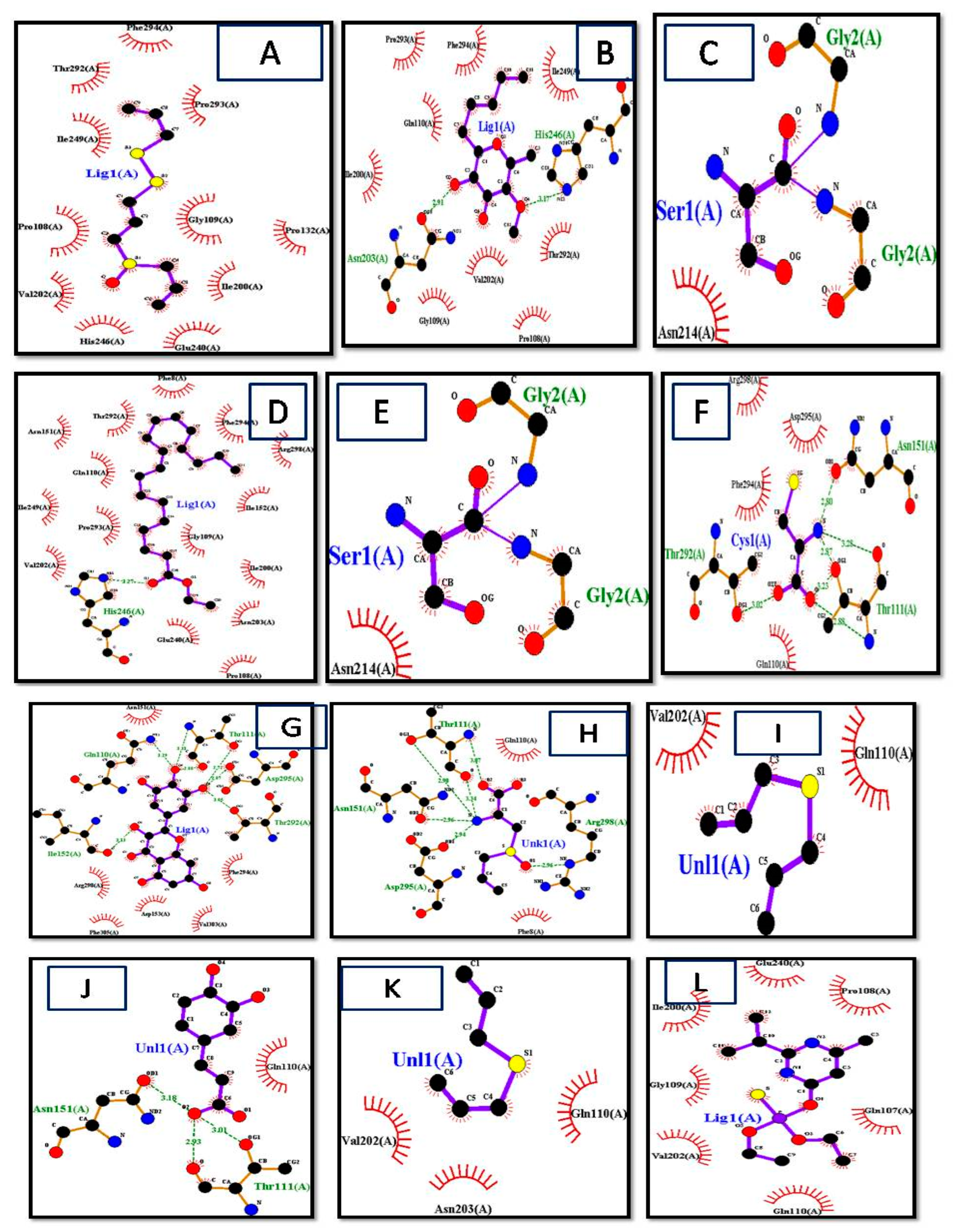
2.9. Visualization of Ligand/Protein
2.10. Analysis of Docked Complex
2.11. Ligand ADMET Properties
2.12. Active Inhibitor Identification
2.13. FDA-Approved Drug-Proposed Antiviral Agent Comparison
3. Results and Discussion
3.1. Target Proteins Structure and Properties
3.2. Ligand Selection and Molecular Docking
3.3. ADMET Properties of Ligands
4. Lead Compounds Identification
5. Drug Identification against COVID-19
6. Reference Drug ADMET Properties
6.1. Absorption Properties
6.2. Distribution Properties
6.3. Metabolic Properties
6.4. Excretion Properties
6.5. Toxicity Prediction of Reference Drug
7. Remdesivir Molecular Docking
8. Remdesivir Comparison with Lead Compound
9. ADMET Properties Comparison
9.1. Absorption Properties Comparison
9.2. Metabolic Properties Comparison
9.3. Distribution Properties Comparison
9.4. Excretion Properties Comparison
9.5. Toxicity Comparison
9.6. Physiochemical Properties Comparison
9.7. Docking Score Comparison
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. COVID-19 Weekly Epidemiological Update, 127th ed.; WHO: Geneva, Switzerland, 2023; 18p. [Google Scholar]
- She, J.; Jiang, J.; Ye, L.; Hu, L.; Bai, C.; Song, Y. 2019 novel coronavirus of pneumonia in Wuhan, China: Emerging attack and management strategies. Clin. Transl. Med. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, A.; Ferrara, F.; Auti, A.M.; Di Domenico, M.; Boccellino, M. Advances in the Omicron variant development. J. Intern. Med. 2022, 292, 81–90. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef] [PubMed]
- WHO. Summary Table of SARS Cases by Country; WHO: Geneva, Switzerland, 2002. [Google Scholar]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203.e2193. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Chang, D.; Cruz, C.S.D. Does inflammation help during COVID-19? ERJ Open Res. 2020, 6, 00557-2020. [Google Scholar] [CrossRef] [PubMed]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef]
- Tomalka, J.A.; Suthar, M.S.; Deeks, S.G.; Sekaly, R.P. Fighting the SARS-CoV-2 pandemic requires a global approach to understanding the heterogeneity of vaccine responses. Nat. Immunol. 2022, 23, 360–370. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Broad-spectrum prodrugs with anti-SARS-CoV-2 activities: Strategies, benefits, and challenges. J. Med. Virol. 2022, 94, 1373–1390. [Google Scholar] [CrossRef]
- Elkarhat, Z.; Charoute, H.; Elkhattabi, L.; Barakat, A.; Rouba, H. Potential inhibitors of SARS-cov-2 RNA dependent RNA polymerase protein: Molecular docking, molecular dynamics simulations and MM-PBSA analyses. J. Biomol. Struct. Dyn. 2022, 40, 361–374. [Google Scholar] [CrossRef]
- van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O. Glycyrrhizin effectively inhibits SARS-CoV-2 replication by inhibiting the viral main protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Jeon, S.; Lee, J.; Kim, S.; Jang, M.S.; Park, C.M.; Song, J.H.; Kim, H.R.; Kwon, S. Broad spectrum antiviral properties of cardiotonic steroids used as potential therapeutics for emerging coronavirus infections. Pharmaceutics 2021, 13, 1839. [Google Scholar] [CrossRef] [PubMed]
- Jahan, I.; Ahmet, O. Potentials of plant-based substance to inhabit and probable cure for the COVID-19. Turk. J. Biol. 2020, 44, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Bhattacharyya, D.; Kasle, G.; Karmakar, S.; Sahu, O.; Ganguly, A.; Addya, S.; Das Sarma, J. Potential immunomodulatory properties of biologically active components of spices against SARS-CoV-2 and Pan β-Coronaviruses. Front. Cell. Infect. Microbiol. 2021, 11, 729622. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Tan, K.-P.; Wang, Y.-M.; Lin, S.-W.; Liang, P.-H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorganic Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Dilshad, E.; Mehmood, F.; Tariq, R.; Munir, A.; Fazal, S. Chemical Constituents of Artemisia Annua are Potent Inhibitors of Alpha-Amylase in Type II Diabetes. Life Sci. 2022, 3, 10. [Google Scholar] [CrossRef]
- Dilshad, E.; Maaz, M.; Ashraf, N.M. Identification of Anti-inflammatory Metabolites from Trigonella foenum-greacum using Computational Approaches. Curr. Trends OMICS 2022, 2, 55–88. [Google Scholar] [CrossRef]
- Eweas, A.F.; Alhossary, A.A.; Abdel-Moneim, A.S. Molecular docking reveals ivermectin and remdesivir as potential repurposed drugs against SARS-CoV-2. Front. Microbiol. 2021, 11, 3602. [Google Scholar] [CrossRef]
- Dilshad, E.; Idrees, M.; Sajid, B.; Munir, A.; Fazal, S. Non-Toxic Flavonoids of Artemisia annua can be used as Anti-Cancer Compounds: A Computational Analysis. Life Sci. 2022, 3, 12. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Razali, R.; Asis, H.; Budiman, C. Structure-function characteristics of SARS-CoV-2 proteases and their potential inhibitors from microbial sources. Microorganisms 2021, 9, 2481. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Li, T. Discovery of alliin as a putative inhibitor of the main protease of SARS-CoV-2 by molecular docking. Biotechniques 2020, 69, 108–112. [Google Scholar] [CrossRef]
- Benhander, G.M.; Abdusalam, A.A.A. Identification of potential inhibitors of SARS-CoV-2 main protease from Allium roseum L. molecular docking study. Chem. Afr. 2022, 5, 57–67. [Google Scholar] [CrossRef]
- Marino, M.; Jamal, Z.; Zito, P.M. Pharmacodynamics. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Malin, J.J.; Suárez, I.; Priesner, V.; Fätkenheuer, G.; Rybniker, J. Remdesivir against COVID-19 and other viral diseases. Clin. Microbiol. Rev. 2020, 34, e00162-20. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Shivanika, C.; Kumar, D.; Ragunathan, V.; Tiwari, P.; Sumitha, A. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020, 40, 585–611. [Google Scholar]
- Wright, S.H. Molecular and cellular physiology of organic cation transporter 2. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1669–F1679. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, D.; Vuković, K.; Toma, C.; Lavado, G.J.; Karmaus, A.L.; Mansouri, K.; Kleinstreuer, N.C.; Benfenati, E.; Roncaglioni, A. SAR and QSAR modeling of a large collection of LD50 rat acute oral toxicity data. J. Cheminformatics 2019, 11, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
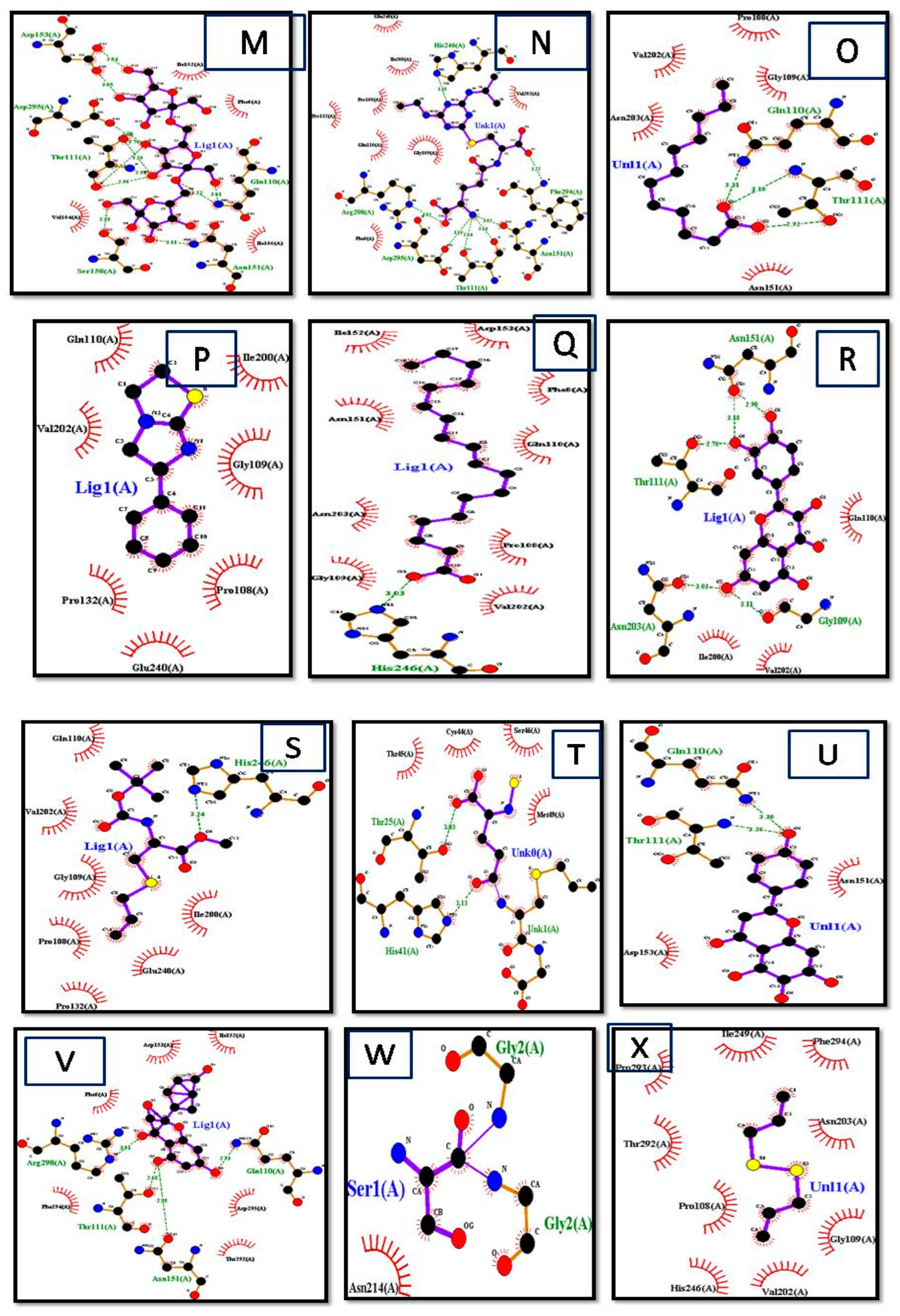{kind=link}
| Pocket ID | Area (SA) | Volume (SA) |
|---|---|---|
| 1 | 284.664 | 292.690 |
| 2 | 273.913 | 214.993 |
| 3 | 53.189 | 59.074 |
| 4 | 104.306 | 30.390 |
| 5 | 40.514 | 25.655 |
| 6 | 27.458 | 7.991 |
| 7 | 20.634 | 7.083 |
| 8 | 13.671 | 4.642 |
| 9 | 6.817 | 3.399 |
| 10 | 16.544 | 3.174 |
| 11 | 10.896 | 2.007 |
| 12 | 10.676 | 1.878 |
| 13 | 6.664 | 1.663 |
| 14 | 6.528 | 0.747 |
| 15 | 4.217 | 0.743 |
| 16 | 6.818 | 0.470 |
| 17 | 4.637 | 0.424 |
| 18 | 4.031 | 0.356 |
| 19 | 1.004 | 0.163 |
| 20 | 1.770 | 0.100 |
| 21 | 0.993 | 0.038 |
| 22 | 0.744 | 0.020 |
| 23 | 0.265 | 0.006 |
| 24 | 0.103 | 0.002 |
| 25 | 0.071 | 0.001 |
| 26 | 0.046 | 0.000 |
| 27 | 0.001 | 0.000 |
| 28 | 0.022 | 0.000 |
| 29 | 0.000 | 0.000 |
| 30 | 0.013 | 0.000 |
| 31 | 0.042 | 0.000 |
| Sr. No | Ligand | Binding Score | Cavity Size | Grid Map | Min-Energy (Kcl/mol) | Max-Energy (Kcl/mol) |
|---|---|---|---|---|---|---|
| 1 | 3-(Allylsulphinyl)-L-Alanine | −4.8 | 1385 | 26 | 0 | 1.6 × 100 |
| 2 | Allicin | −3.2 | 1385 | 26 | 0 | 1.6 × 100 |
| 3 | Diallyl Sulfide | −3.1 | 1385 | 26 | 0 | 1.6 × 100 |
| 4 | Diallyl Disulfide | −3.5 | 1385 | 26 | 0 | 1.6 × 100 |
| 5 | Diallyl Trisulfide | −5.5 | 277 | 21 | 0 | 1.6 × 100 |
| 6 | Glutathione | −5.8 | 277 | 21 | 0 | 1.6 × 100 |
| 7 | L-Cysteine | −3.7 | 1385 | 26 | 0 | 1.6 × 100 |
| 8 | S-Allyl-Mercapto-Glutathione | −6 | 277 | 22 | 0 | 1.6 × 100 |
| 9 | Thiocysteine | −3.8 | 1385 | 21 | 0 | 1.6 × 100 |
| 10 | Gamma-Glutamyl-L-Cysteine | −7.2 | 1385 | 24 | 0 | 1.6 × 100 |
| 11 | Gamma-Glutamyl-S-Allylcysteine | −5.4 | 1385 | 21 | 0 | 1.6 × 100 |
| 12 | Kaempferol | −7.4 | 1385 | 26 | 0 | 1.6 × 100 |
| 13 | Quercetin | −7.6 | 1385 | 21 | 0 | 1.6 × 100 |
| 14 | Myricetin | −7.8 | 1385 | 21 | 0 | 1.6 × 100 |
| 15 | Fructan | −7.1 | 1385 | 21 | 0 | 1.6 × 100 |
| 16 | Lauric Acid | −5.2 | 1385 | 22 | 0 | 1.6 × 100 |
| 17 | Linoleic Acid | −5.7 | 1385 | 30 | 0 | 1.6 × 100 |
| 18 | Allixin | −5.8 | 1385 | 26 | 0 | 1.6 × 100 |
| 19 | Ajoene | −4.7 | 1385 | 22 | 0 | 1.6 × 100 |
| 20 | Ethyl Linoleate | −5.8 | 1385 | 31 | 0 | 1.6 × 100 |
| 21 | Diazinon | −5.7 | 1385 | 26 | 0 | 1.6 × 100 |
| 22 | Levamisole | −5.7 | 1385 | 26 | 0 | 1.6 × 100 |
| 23 | Scutellarein | −7.6 | 1385 | 21 | 0 | 1.6 × 100 |
| 24 | S-allyl cysteine methyl ester | −5.3 | 1385 | 21 | 0 | 1.6 × 100 |
| 25 | Caffeic acid | −5.8 | 1385 | 26 | 0 | 1.6 × 100 |
| Sr. No | Ligand | LogP | Molecular Weight(g/mol) | Hydrogen Bond Acceptor | Hydrogen Bond Donar |
|---|---|---|---|---|---|
| 1 | L-Alanine | −0.667 | 177.225 | 3 | 2 |
| 2 | Allicin | 1.7553 | 162.279 | 2 | 0 |
| 3 | Diallyl Sulfide | 2.0916 | 114.213 | 1 | 0 |
| 4 | Diallyl Disulfide | 2.7398 | 146.28 | 2 | 0 |
| 5 | Diallyl Trisulfide | 3.388 | 178.347 | 3 | 0 |
| 6 | Glutathione | −2.2061 | 307.328 | 6 | 6 |
| 7 | L-Cysteine | −0.6719 | 121.161 | 3 | 3 |
| 8 | S-Allyl-Mercapto- Glutathione | −0.741 | 379.46 | 7 | 6 |
| 9 | Thiocysteine | −0.0237 | 153.228 | 4 | 3 |
| 10 | L-Cysteine | l- −0.0227 | 429.503 | l- 10 10 | 6 |
| 11 | Gamma-Glutamyl- - S-Allylcysteine | −3329 | 290.341 | 5 | 4 |
| 12 | Kaempferol | 2.2824 | 286.239 | 6 | 4 |
| 13 | Quercetin | 1.988 | 302.238 | 7 | 5 |
| 14 | Myricetin | 1.6936 | 318.237 | 8 | 6 |
| 15 | Fructan | −7.5682 | 504.438 | 16 | 11 |
| 16 | Lauric Acid | 3.9919 | 200.322 | 1 | 1 |
| 17 | Linoleic Acid | 5.8845 | 280.452 | 1 | 1 |
| 18 | Allixin | 2.39512 | 226.272 | 4 | 1 |
| 19 | Ajoene | 3.0022 | 234.411 | 3 | 0 |
| 20 | Ethyl Linoleate | 6.363 | 308.506 | 2 | 0 |
| 21 | Diazinon | 3.58472 | 304.352 | 6 | 0 |
| 22 | Levamisole | 2.1461 | 204.298 | 3 | 0 |
| 23 | Scutellarein | 2.2824 | 286.239 | 6 | 4 |
| 24 | S-allyl cysteine methyl ester | 1.9719 | 275.37 | 5 | 1 |
| 25 | Caffeic acid | 2.987 | 180.159 | 3 | 3 |
| Sr. No | Ligand | Water Solubility | Caco2 Permeability | Intestinal Absorption (Human) | Skin Permeability | P-Glucoprotein Substrate | P-Glucoprotein I inhibitor | P-glucoprotein II Inhibitor |
|---|---|---|---|---|---|---|---|---|
| 1 | -L-Alanine | −2.888 | 0.619 | 76.495 | −2.735 | No | No | No |
| 2 | Allicin | −1.72 | 1.316 | 96.229 | −1.877 | No | No | No |
| 3 | Diallyl Sulfide | −2.695 | 1.394 | 96.268 | −1.488 | No | No | No |
| 4 | Diallyl Disulfide | −3.222 | 1.399 | 94.769 | −1.429 | No | No | No |
| 5 | Diallyl Trisulfide | −3.781 | 1.403 | 92.573 | −1.449 | No | No | No |
| 6 | Glutathione | −2.892 | −0.536 | 0 | −2.735 | Yes | No | No |
| 7 | L-Cysteine | −2.888 | 0.386 | 74.807 | −2.737 | No | No | No |
| 8 | S-Allyl-Mercapto- Glutathione | −2.205 | −0.457 | 0 | −2.735 | Yes | No | No |
| 9 | Thiocysteine | −2.887 | 0.424 | 78.653 | −2.737 | No | No | No |
| 10 | Gamma-Glutamyl- L-Cysteine | −2.892 | −0.598 | 0.259 | −2.735 | Yes | No | No |
| 11 | Gamma-Glutamyl- S-Allylcysteine | −2.891 | −0.517 | 8.312 | −2.735 | Yes | No | No |
| 12 | Kaempferol | −3.04 | 0.032 | 74.29 | −2.735 | Yes | No | No |
| 13 | Quercetin | −2.925 | −0.229 | 77.207 | −2.735 | Yes | No | No |
| 14 | Myricetin | −2.915 | 0.095 | 65.93 | −2.735 | Yes | No | No |
| 15 | Fructan | −1.2 | −0.835 | 0 | −2.735 | Yes | No | No |
| 16 | Lauric Acid | −4.181 | 1.562 | 93.379 | −2.693 | No | No | No |
| 17 | Linoleic Acid | −5.862 | 1.57 | 92.329 | −2.723 | No | No | No |
| 18 | Allixin | −3.074 | 1.301 | 93.438 | −3.141 | No | No | No |
| 19 | Ajoene | −3.54 | 1.329 | 95.186 | −1.745 | No | No | No |
| 20 | Ethyl Linoleate | −7.525 | 1.608 | 92.241 | −2.774 | No | No | Yes |
| 21 | Diazinon | −3.757 | 1.509 | 92.749 | −3.005 | No | No | No |
| 22 | Levamisole | −3.173 | 1.491 | 93.678 | −2.075 | No | No | No |
| 23 | Scutellarein | −3.156 | −0.357 | 66.687 | −2.735 | Yes | No | No |
| 24 | S-allylcysteine methylester | −2.213 | 0.986 | 93.247 | −3.061 | No | No | No |
| 25 | Caffeic acid | −2.33 | 0.634 | 69.407 | −2.722 | No | No | No |
| Sr. No | Ligand | VDss (Human) | Fraction Unbound (Human) | BBB Permeability (Human) | CNS Permeability |
|---|---|---|---|---|---|
| 1 | -L-Alanine | −0.553 | 0.462 | −0.271 | −3.472 |
| 2 | Allicin | −0.045 | 0.577 | 0.506 | −2.312 |
| 3 | Diallyl Sulfide | 0.202 | 0.552 | 0.69 | −2.102 |
| 4 | Diallyl Disulfide | 0.211 | 0.518 | 0.78 | −2.21 |
| 5 | Diallyl Trisulfide | 0.216 | 0.483 | 0.767 | −2.309 |
| 6 | Glutathione | −0.377 | 0.463 | −1.085 | −3.903 |
| 7 | L-Cysteine | −0.486 | 0.49 | −0.398 | −3.476 |
| 8 | S-Allyl-Mercapto- Glutathione | −1.517 | 0.588 | −1.475 | −4.217 |
| 9 | Thiocysteine | −0.501 | 0.47 | −0.376 | −3.5 |
| 10 | Gamma-Glutamyl- L-Cysteine | −0.203 | 0.495 | −1.994 | −4.159 |
| 11 | Gamma-Glutamyl- S-Allylcysteine | −0.48 | 0.452 | −1.124 | −4.02 |
| 12 | Kaempferol | 1.274 | 0.178 | −0.939 | −2.228 |
| 13 | Quercetin | 1.559 | 0.206 | −1.098 | −3.065 |
| 14 | Myricetin | 1.317 | 0.238 | −1.493 | −3.709 |
| 15 | Fructan | −0.276 | 0.499 | −1.886 | −4.815 |
| 16 | Lauric Acid | −0.631 | 0.26 | 0.057 | −2.034 |
| 17 | Linoleic Acid | −0.587 | 0.054 | −0.142 | −1.6 |
| 18 | Allixin | −0.008 | 0.479 | 0.193 | −2.86 |
| 19 | Ajoene | 0.083 | 0.395 | 0.703 | −2.178 |
| 20 | Ethyl Linoleate | 0.306 | 0.015 | 0.776 | −1.562 |
| 21 | Diazinon | −0.348 | 0.329 | −0.438 | −3.029 |
| 22 | Levamisole | 0.428 | 0.358 | 0.358 | −2.011 |
| 23 | Scutellarein | 0.587 | 0.192 | −1.398 | −2.363 |
| 24 | S-allylcysteine methylester | -0.396 | 0.434 | −0.119 | −2.911 |
| 25 | Caffeic acid | -1.098 | 0.529 | −0.647 | −2.608 |
| Sr. No | Ligands | CYP- 2D6 Substrate | CYP- 3A4 Substrate | CYP- 2D6 Inhibitor | CYP- 2C19 Inhibitor | CYP- 2C9 Inhibitor | CYP- 2D6 Inhibitor | CYP- 3A4 Inhibitor |
|---|---|---|---|---|---|---|---|---|
| 1 | -L-Alanine | NO | NO | NO | NO | NO | NO | NO |
| 2 | Allicin | NO | NO | NO | NO | NO | NO | NO |
| 3 | Diallyl Sulfide | NO | NO | NO | NO | NO | NO | NO |
| 4 | Diallyl Disulfide | NO | NO | NO | NO | NO | NO | NO |
| 5 | Diallyl Trisulfide | NO | NO | NO | NO | NO | NO | NO |
| 6 | Glutathione | NO | NO | NO | NO | NO | NO | NO |
| 7 | L-Cysteine | NO | NO | NO | NO | NO | NO | NO |
| 8 | S-Allyl-Mercapto- Glutathione | NO | NO | NO | NO | NO | NO | NO |
| 9 | Thiocysteine | NO | NO | NO | NO | NO | NO | NO |
| 10 | Gamma-Glutamyl- L-Cysteine | NO | NO | NO | NO | NO | NO | NO |
| 11 | Gamma-Glutamyl- S-Allylcysteine | NO | NO | NO | NO | NO | NO | NO |
| 12 | Kaempferol | NO | NO | YES | NO | NO | NO | NO |
| 13 | Quercetin | NO | NO | YES | NO | NO | NO | NO |
| 14 | Myricetin | NO | NO | YES | NO | NO | NO | NO |
| 15 | Fructan | NO | NO | NO | NO | NO | NO | NO |
| 16 | Lauric Acid | NO | NO | NO | NO | NO | NO | NO |
| 17 | Linoleic Acid | NO | YES | YES | NO | NO | NO | NO |
| 18 | Allixin | NO | NO | YES | NO | NO | NO | NO |
| 19 | Ajoene | NO | NO | NO | NO | NO | NO | NO |
| 20 | EthylLinoleate | NO | YES | YES | NO | NO | NO | NO |
| 21 | Diazinon | NO | NO | NO | NO | NO | NO | YES |
| 22 | Levamisole | NO | NO | YES | NO | NO | NO | NO |
| 23 | Scutellarein | NO | NO | YES | NO | NO | YES | NO |
| 24 | S-allylcysteine methylester | NO | NO | NO | NO | NO | NO | NO |
| 25 | Caffeic acid | NO | NO | NO | NO | NO | NO | NO |
| Sr. No | Ligands | Total Clearance | Renal OCT2 Substrate |
|---|---|---|---|
| 1 | -L-Alanine | 0.365 | No |
| 2 | Allicin | 0.714 | No |
| 3 | DiallylSulfide | 0.555 | No |
| 4 | DiallylDisulfide | 0.547 | No |
| 5 | DiallylTrisulfide | 0.446 | No |
| 6 | Glutathione | 0.308 | No |
| 7 | L-Cysteine | 0.53 | No |
| 8 | S-Allyl-Mercapto- Glutathione | 0.333 | No |
| 9 | Thiocysteine | 0.369 | No |
| 10 | Gamma-Glutamyl- L-Cysteine | Gamma-Glutamyl- 0.159 L-Cysteine | No |
| 11 | Gamma-Glutamyl- S-Allylcysteine | - 0.3 | No |
| 12 | Kaempferol | 0.477 | No |
| 13 | Quercetin | 0.407 | No |
| 14 | Myricetin | 0.422 | No |
| 15 | Fructan | 1.516 | No |
| 16 | Lauric Acid | 1.623 | No |
| 17 | Linoleic Acid | 1.936 | No |
| 18 | Allixin | 0.419 | No |
| 19 | Ajoene | 0.538 | No |
| 20 | Ethyl Linoleate | 2.08 | No |
| 21 | Diazinon | 0.391 | No |
| 22 | Levamisole | 0.475 | No |
| 23 | Scutellarein | 0.47 | No |
| 24 | S-allylcysteine Methylester | 0.487 | No |
| 25 | Caffeic acid | 0.508 | No |
| Ligands | Max. Tolerated Dose (Human) mg/Kg | hERG I inhibitor | hERGII inhibitor | Oral Rat Acute Toxicity | Oral Rat Chronic Toxicity | Hepa toxicity | Skin Sensitization | t.Pyriformis Toxicity | Minnow Toxicity |
|---|---|---|---|---|---|---|---|---|---|
| -L-Alanine | 1.164 | No | No | 2.051 | 1.9 | No | No | 0.268 | 2.598 |
| Allicin | 0.737 | No | No | 2.366 | 1.406 | No | Yes | 0.9 | 1.235 |
| DiallylSulfide | 0.782 | No | No | 2.028 | 1.812 | No | Yes | 0.63 | 1.154 |
| DiallylDisulfide | 0.674 | No | No | 2.375 | 1.847 | No | Yes | 1.371 | 0.79 |
| DiallylTrisulfide | 0.582 | No | No | 2.711 | 1.8 57 | No | Yes | 2.008 | 0.516 |
| Glutathione | 1.104 | No | No | 2.468 | 2.919 | NO | NO | 0.285 | 4.569 |
| L-Cysteine | 1.133 | No | No | 1.982 | 2.6 | NO | NO | 0.149 | 2.992 |
| S-Allyl-Mercapto- Glutathione | 1.196 | No | No | 1.804 | 2.902 | YES | NO | 0.285 | 4.569 |
| Thiocysteine | 1.113 | No | No | 1.983 | 2.275 | NO | NO | 0.149 | 2.992 |
| Gamma-Glutamyl- L-Cysteine | 0.856 | No | No | 2.478 | 3.361 | YES | NO | 0.285 | 4.164 |
| Gamma-Glutamyl- S-Allylcysteine | 1.119 | No | No | 2.438 | 3.361 | NO | NO | 0.101 | 2.657 |
| Kaempferol | 0.531 | No | No | 2.449 | 2.29 | NO | NO | 0.285 | 4.306 |
| Quercetin | 0.499 | No | No | 2.471 | 2.505 | NO | NO | 0.285 | 2.928 |
| Myricetin | 0.51 | No | No | 2.497 | 2.618 | NO | NO | 0.314 | 2.885 |
| Fructan | 0.667 | No | No | 2.775 | 2.718 | NO | NO | 0.288 | 3.721 |
| Lauric Acid | −0.34 | No | No | 1.511 | 4.703 | NO | NO | 0.286 | 5.023 |
| Linoleic Acid | −8.27 | No | No | 1.429 | 2.89 | NO | NO | 0.285 | 13.29 |
| Allixin | −0.879 | No | No | 2.195 | 3.187 | NO | NO | 0.945 | −0.084 |
| Ajoene | 0.462 | No | No | 2.472 | 0.899 | NO | YES | 0.701 | −1.31 |
| EthylLinoleate | 0.009 | No | No | 1.644 | 3.023 | NO | YES | 0.324 | 1.582 |
| Diazinon | 1.362 | No | No | 3.258 | 0.953 | YES | NO | 2.197 | 0.155 |
| Levamisole | 0.035 | No | No | 2.711 | 1.548 | NO | YES | 1.497 | −1.765 |
| Scutellarein | 0.626 | No | No | 2.452 | 3.135 | NO | NO | 0.366 | −0.148 |
| S-allylcysteine methylester | 0.703 | No | No | 2.6 | 0.908 | NO | NO | 1.355 | 1.45 |
| Caffeic acid | 1.145 | No | No | 2.383 | 2.092 | NO | NO | 0.301 | 1.99 |
| Ligands | Water Solubility | CaCO2 Permeability | Intestinal Absorption (human) | Skin Permeability | P-Gluco Protein Substrate | P-Gluco Protein I Inhibitor | P-Gluco Protein II Inhibitor |
|---|---|---|---|---|---|---|---|
| Remdesivir | −3.07 | 0.635 | 71.109 | 2.735 | Yes | Yes | No |
| Ligand | VDss (Human) | Fraction Unbound (Human) | BBB Permeability (Human) | CNS Permeability |
|---|---|---|---|---|
| Remdesivir | 0.307 | 0.005 | −2.056 | −4.675 |
| Ligand | CYP2D6 Substrate | CYP3A4 Substrate | CYP2D6 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|
| Remdesivir | NO | YES | NO | NO | NO | NO | NO |
| Ligands | Total Clearance | Renal OCT2 Substrate |
|---|---|---|
| Remdesivir | 0.198 | NO |
| Ligands | Max. Tolerated Dose (Human) | Herg I Inhibitor | Herg II Inhibitor | Oral Rat Acute Toxicity | Oral Rat Chronic Toxicity | Hepatoxicity | Skin Sensitization | T.Pyriformis Toxicity | Minnow Toxicity (Log Mm) |
|---|---|---|---|---|---|---|---|---|---|
| Remdesivir | 1.972 | No | Yes | 2.043 | 1.639 | Yes | No | 0.285 | 0.291 |
| Ligands | Binding Score | Cavity Size | Grid Map | HBA | HBD | logP | Mol. Weight g/mol |
|---|---|---|---|---|---|---|---|
| Remdesivir | −8.1 | 1385 | 22 | 13 | 4 | 2.31218 | 602.585 |
| Ligand | LogP | Molecular Weight (g/mol) | Hydrogen Bond Acceptor | Hydrogen Bond Donar |
|---|---|---|---|---|
| Remdesivir | 2.31218 | 602.585 | 13 | 4 |
| Levamisole | 2.1461 | 204.29 | 3 | 0 |
| Ligands | Water Solubility | CaCO2 Permeability | Intestinal Absorption (Human) | Skin Permeability | P-Glucoprotein Substrate | P-Glucoprotein I Inhibitor | PglucoproteinII Inhibitor |
|---|---|---|---|---|---|---|---|
| Remdesivir | −3.07 | 0.635 | 71.109 | −2.735 | Yes | Yes | No |
| Levamisole | −3.173 | 1.491 | 93.678 | −2.075 | No | No | No |
| Ligand | CYP2D6 Substrate | CYP3A4 Substrate | CYP2D6 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP2D6 Inhibitor |
|---|---|---|---|---|---|---|---|
| Remdesivir | No | Yes | No | No | No | No | No |
| Levamisole | No | No | Yes | No | No | Yes | No |
| Ligands | VDss (Human) | Fraction Unbound (Human) | BBB Permeability (Human) | CNS Permeability |
|---|---|---|---|---|
| Remdesivir | 0.307 | 0.005 | −2.056 | −4.675 |
| Levamisole | 0.428 | 0.358 | 0.358 | −2.011 |
| Ligands | Total Clearance | Renal OCT2 Substrate |
|---|---|---|
| Remdesivir | 0.198 | No |
| Levamisole | 0.475 | No |
| Ligand | Max. Tolerated Dose (Human) (mg/Kg) | HergI Inhibitor | HergII Inhibitor | Oral Rat Acute Toxicity (mol/Kg) | Oral Rat Chronic Toxicity (mol/Kg) | Hepa Toxicity | Skin Sensitization | T.Pyriforms Toxicity (LogUg/L) | Minnow Toxicity (Log Mm) |
|---|---|---|---|---|---|---|---|---|---|
| Remdesivir | 0.196 | No | Yes | 2.043 | 1.639 | Yes | No | 0.285 | 0.291 |
| Levamisole | 0.035 | No | No | 2.711 | 1.548 | No | Yes | 1.355 | 1.45 |
| Ligands | LogP | Molecular Weight (g/mol) | Molecular Formula | HBond Acceptor | HBond Donar |
|---|---|---|---|---|---|
| Remdesivir | 2.31218 | 602.585 | C27H35N6O8P | 13 | 4 |
| Levamisole | 2.1461 | 204.29 | C11H12N2S | 3 | 0 |
| Ligands | Score |
|---|---|
| Remdesivir | −8.1 |
| Levamisole | −5.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, H.; Dilshad, E.; Afsar, T.; Almajwal, A.; Shafique, H.; Razak, S. Molecular Screening of Bioactive Compounds of Garlic for Therapeutic Effects against COVID-19. Biomedicines 2023, 11, 643. https://doi.org/10.3390/biomedicines11020643
Ashraf H, Dilshad E, Afsar T, Almajwal A, Shafique H, Razak S. Molecular Screening of Bioactive Compounds of Garlic for Therapeutic Effects against COVID-19. Biomedicines. 2023; 11(2):643. https://doi.org/10.3390/biomedicines11020643
Chicago/Turabian StyleAshraf, Huma, Erum Dilshad, Tayyaba Afsar, Ali Almajwal, Huma Shafique, and Suhail Razak. 2023. "Molecular Screening of Bioactive Compounds of Garlic for Therapeutic Effects against COVID-19" Biomedicines 11, no. 2: 643. https://doi.org/10.3390/biomedicines11020643
APA StyleAshraf, H., Dilshad, E., Afsar, T., Almajwal, A., Shafique, H., & Razak, S. (2023). Molecular Screening of Bioactive Compounds of Garlic for Therapeutic Effects against COVID-19. Biomedicines, 11(2), 643. https://doi.org/10.3390/biomedicines11020643