

Mechanotransduction Impairment in Primary Fibroblast Model of Krabbe Disease

 ,
,  ,
,  , ,
, , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals and Cells
2.2. Cell Culture, Drugs’ Treatments, and Cell Viability Assay
2.3. Single-Cell Migration
2.4. Wound Healing
2.5. Immunostaining
2.6. Confocal Fluorescence Microscopy
2.7. FA, NBR1, N-CAD, and YAP Image Analysis
2.8. Western Blot
2.9. Statistical Analysis
3. Results
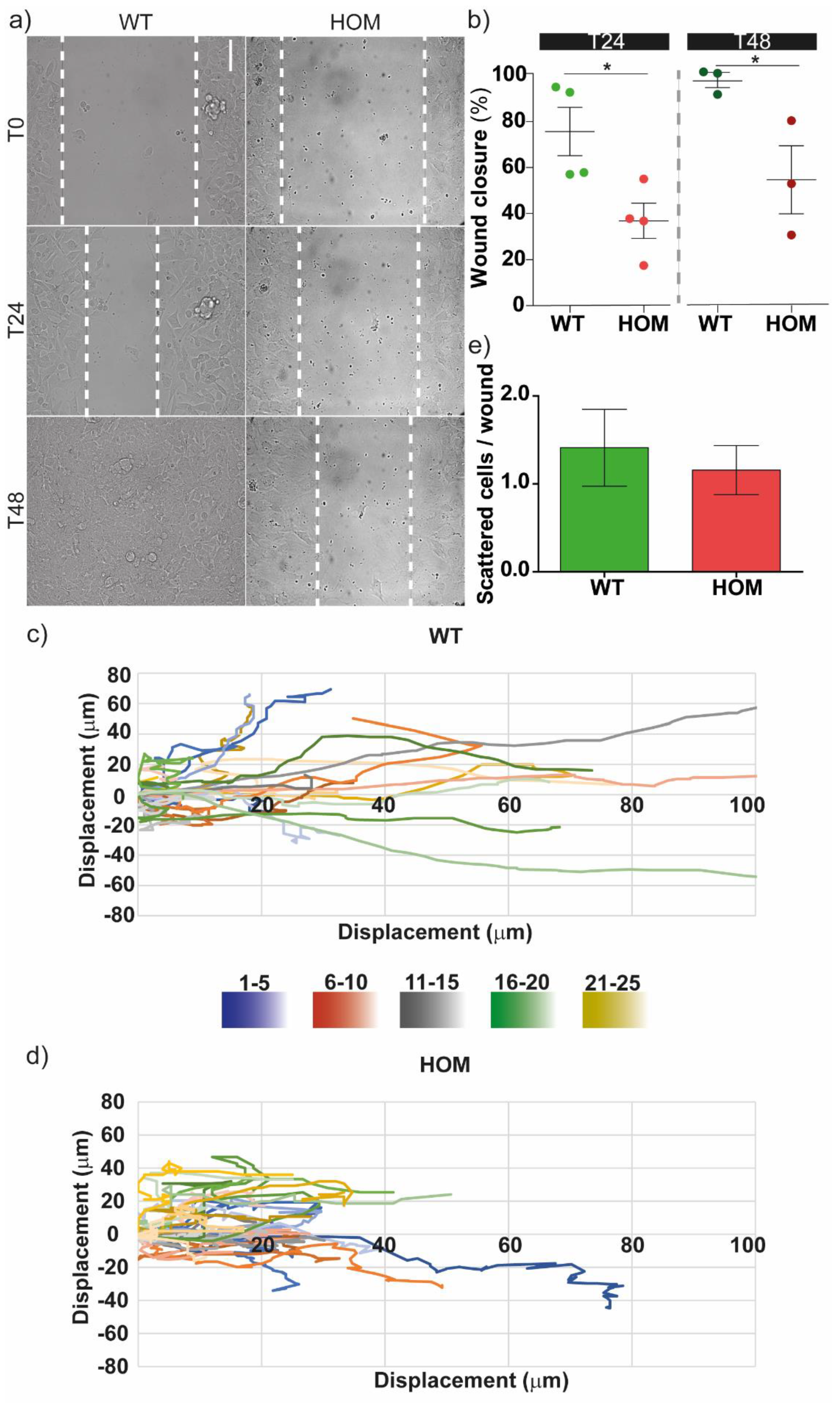
3.1. GALC-Deficient Fibroblasts Showed Impaired Migration at the Single-Cell Level and in Monolayer Wound Healing
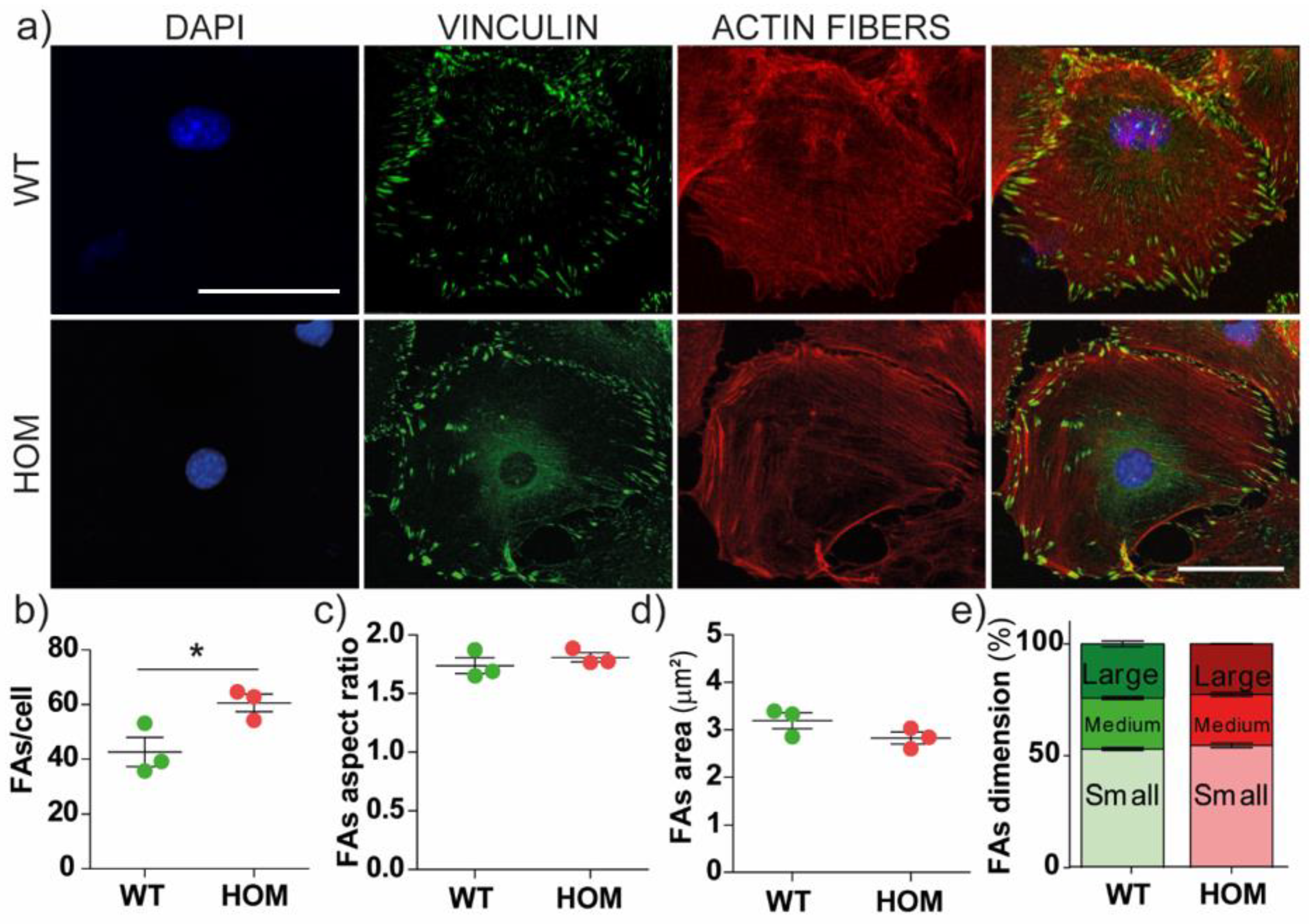
3.2. The Number of Focal Adhesions Increases in GALC-Deficient Fibroblasts
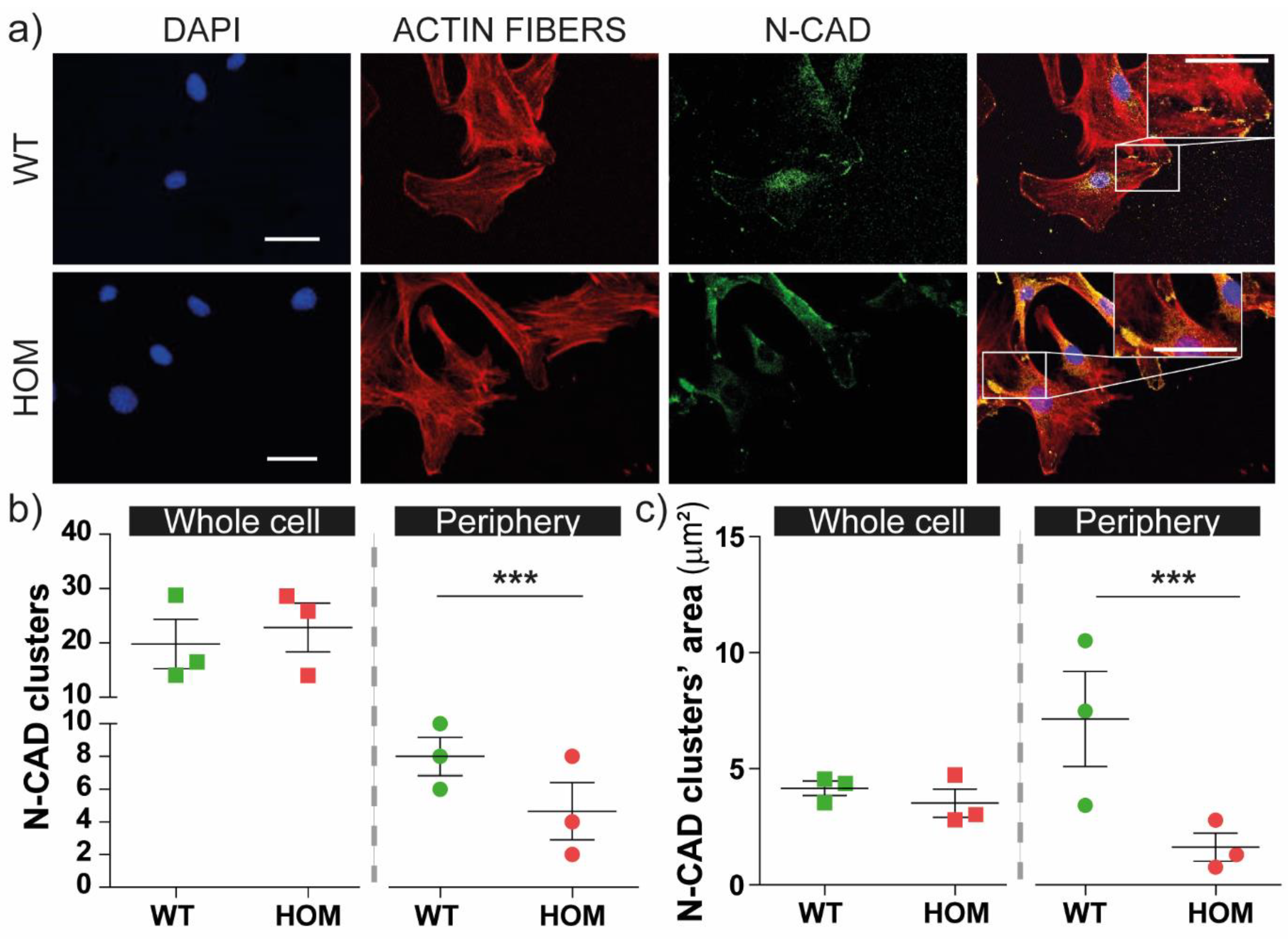
3.3. Dysregulated Distribution and Dimension of N-CAD Junctions
3.4. YAP Mechanosensing Activation
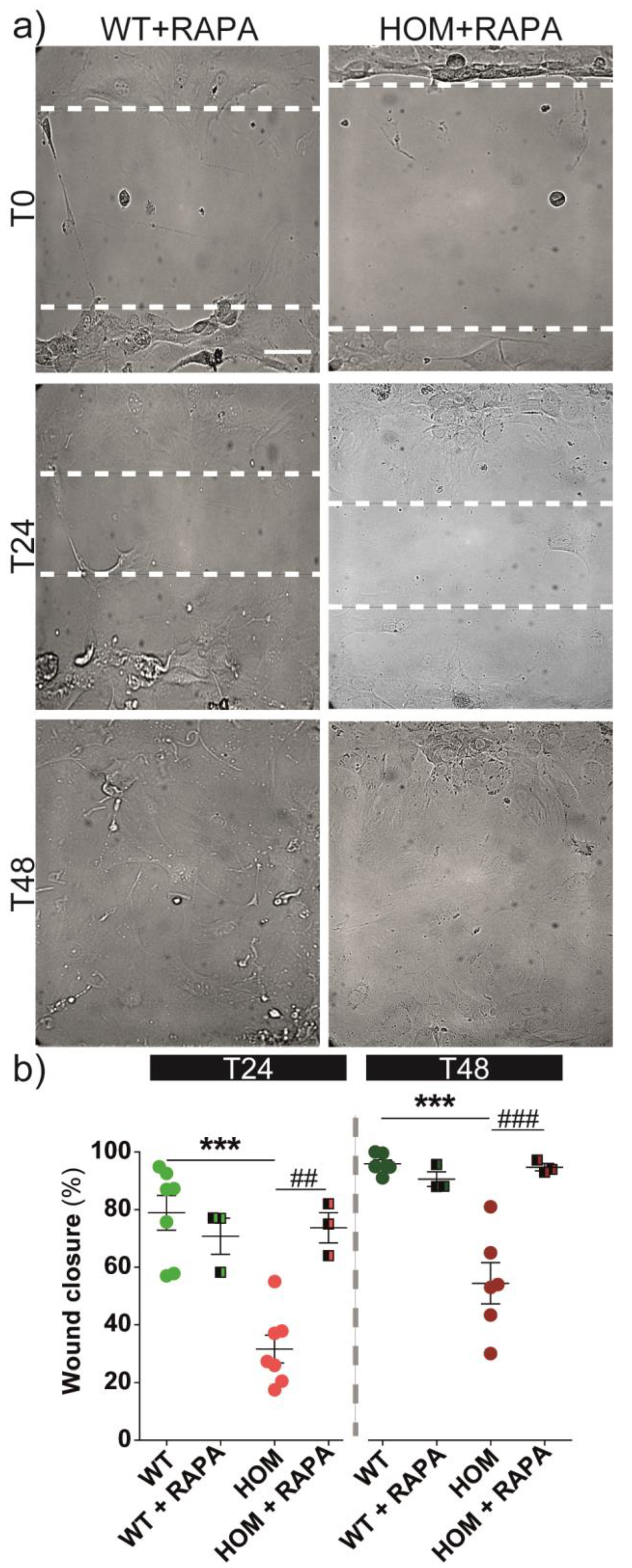
3.5. Autophagy Activation Rescues Cell Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miyatake, T.; Suzuki, K. Globoid Cell Leukodistrophy: Additional Deficiency of Psychosine Galactosidase. Biochem. Biophys. Res. Commun. 1972, 48, 539–543. [Google Scholar] [CrossRef] [PubMed]
- White, A.B.; Givogri, M.I.; Lopez-Rosas, A.; Cao, H.; van Breemen, R.; Thinakaran, G.; Bongarzone, E.R. Psychosine Accumulates in Membrane Microdomains in the Brain of Krabbe Patients, Disrupting the Raft Architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef] [Green Version]
- Hawkins-Salsbury, J.A.; Parameswar, A.R.; Jiang, X.; Schlesinger, P.H.; Bongarzone, E.; Ory, D.S.; Demchenko, A.V.; Sands, M.S. Psychosine, the Cytotoxic Sphingolipid That Accumulates in Globoid Cell Leukodystrophy, Alters Membrane Architecture. J. Lipid Res. 2013, 54, 3303–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Auria, L.; Reiter, C.; Ward, E.; Moyano, A.L.; Marshall, M.S.; Nguyen, D.; Scesa, G.; Hauck, Z.; van Breemen, R.; Givogri, M.I.; et al. Psychosine Enhances the Shedding of Membrane Microvesicles: Implications in Demyelination in Krabbe’s Disease. PLoS ONE 2017, 12, e0178103. [Google Scholar] [CrossRef] [Green Version]
- White, A.B.; Galbiati, F.; Givogri, M.I.; Lopez Rosas, A.; Qiu, X.; van Breemen, R.; Bongarzone, E.R. Persistence of Psychosine in Brain Lipid Rafts Is a Limiting Factor in the Therapeutic Recovery of a Mouse Model for Krabbe Disease. J. Neurosci. Res. 2011, 89, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.-S.; Singh, A.K.; Singh, I. Biochemical, Cell Biological, Pathological, and Therapeutic Aspects of Krabbe’s Disease: Krabbe’s Disease and Therapeutics. J. Neurosci. Res. 2016, 94, 990–1006. [Google Scholar] [CrossRef] [Green Version]
- Misslin, C.; Velasco-Estevez, M.; Albert, M.; O’Sullivan, S.A.; Dev, K.K. Phospholipase A2 Is Involved in Galactosylsphingosine-Induced Astrocyte Toxicity, Neuronal Damage and Demyelination. PLoS ONE 2017, 12, e0187217. [Google Scholar] [CrossRef] [Green Version]
- Won, J.-S.; Kim, J.; Paintlia, M.K.; Singh, I.; Singh, A.K. Role of Endogenous Psychosine Accumulation in Oligodendrocyte Differentiation and Survival: Implication for Krabbe Disease. Brain Res. 2013, 1508, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Ribbens, J.J.; Moser, A.B.; Hubbard, W.C.; Bongarzone, E.R.; Maegawa, G.H.B. Characterization and Application of a Disease-Cell Model for a Neurodegenerative Lysosomal Disease. Mol. Genet. Metab. 2014, 111, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Ijichi, K.; Brown, G.D.; Moore, C.S.; Lee, J.-P.; Winokur, P.N.; Pagarigan, R.; Snyder, E.Y.; Bongarzone, E.R.; Crocker, S.J. MMP-3 Mediates Psychosine-Induced Globoid Cell Formation: Implications for Leukodystrophy Pathology: MMP-3 in Leukodystrophy Pathology. Glia 2013, 61, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Snook, E.R.; Fisher-Perkins, J.M.; Sansing, H.A.; Lee, K.M.; Alvarez, X.; MacLean, A.G.; Peterson, K.E.; Lackner, A.A.; Bunnell, B.A. Innate Immune Activation in the Pathogenesis of a Murine Model of Globoid Cell Leukodystrophy. Am. J. Pathol. 2014, 184, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Voccoli, V.; Tonazzini, I.; Signore, G.; Caleo, M.; Cecchini, M. Role of Extracellular Calcium and Mitochondrial Oxygen Species in Psychosine-Induced Oligodendrocyte Cell Death. Cell Death Dis. 2014, 5, e1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, C.R.; Rebiai, R.; Kwak, A.; Marshall, J.; Wozniak, D.; Scesa, G.; Nguyen, D.; Rue, E.; Pathmasiri, K.C.; Pijewski, R.; et al. The Pathogenic Sphingolipid Psychosine Is Secreted in Extracellular Vesicles in the Brain of a Mouse Model of Krabbe Disease. ASN Neuro 2022, 14, 175909142210878. [Google Scholar] [CrossRef] [PubMed]
- Belleri, M.; Presta, M. Endothelial Cell Dysfunction in Globoid Cell Leukodystrophy: Microvascular Alterations in GLD. J. Neurosci. Res. 2016, 94, 1359–1367. [Google Scholar] [CrossRef]
- Zizioli, D.; Guarienti, M.; Tobia, C.; Gariano, G.; Borsani, G.; Bresciani, R.; Ronca, R.; Giacopuzzi, E.; Preti, A.; Gaudenzi, G.; et al. Molecular Cloning and Knockdown of Galactocerebrosidase in Zebrafish: New Insights into the Pathogenesis of Krabbe’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belleri, M.; Ronca, R.; Coltrini, D.; Nico, B.; Ribatti, D.; Poliani, P.L.; Giacomini, A.; Alessi, P.; Marchesini, S.; Santos, M.B.; et al. Inhibition of Angiogenesis by β-Galactosylceramidase Deficiency in Globoid Cell Leukodystrophy. Brain 2013, 136, 2859–2875. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.A.; Miranda, C.O.; Sousa, V.F.; Santos, T.E.; Malheiro, A.R.; Solomon, M.; Maegawa, G.H.; Brites, P.; Sousa, M.M. Early Axonal Loss Accompanied by Impaired Endocytosis, Abnormal Axonal Transport, and Decreased Microtubule Stability Occur in the Model of Krabbe’s Disease. Neurobiol. Dis. 2014, 66, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Papini, N.; Giallanza, C.; Brioschi, L.; Ranieri, F.R.; Giussani, P.; Mauri, L.; Ciampa, M.G.; Viani, P.; Tringali, C. Galactocerebrosidase deficiency induces an increase in lactosylceramide content: A new hallmark of Krabbe disease? Int. J. Biochem. Cell Biol. 2022, 145, 106184. [Google Scholar] [CrossRef]
- Del Grosso, A.; Angella, L.; Tonazzini, I.; Moscardini, A.; Giordano, N.; Caleo, M.; Rocchiccioli, S.; Cecchini, M. Dysregulated Autophagy as a New Aspect of the Molecular Pathogenesis of Krabbe Disease. Neurobiol. Dis. 2019, 129, 195–207. [Google Scholar] [CrossRef]
- Mangiameli, E.; Cecchele, A.; Morena, F.; Sanvito, F.; Matafora, V.; Cattaneo, A.; della Volpe, L.; Gnani, D.; Paulis, M.; Susani, L.; et al. Human IPSC-Based Neurodevelopmental Models of Globoid Cell Leukodystrophy Uncover Patient- and Cell Type-Specific Disease Phenotypes. Stem Cell Rep. 2021, 16, 1478–1495. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of MTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Kondo, S.; Zhu, W.; Xie, W.; Jankovic, J.; Le, W. Neuroprotection of Rapamycin in Lactacystin-Induced Neurodegeneration via Autophagy Enhancement. Neurobiol. Dis. 2008, 32, 16–25. [Google Scholar] [CrossRef]
- Caramés, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy Activation by Rapamycin Reduces Severity of Experimental Osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.A.; Rao, H.Z.; Luzi, P.; Wenger, D.A. Long-Term Improvements in Lifespan and Pathology in CNS and PNS After BMT Plus One Intravenous Injection of AAVrh10-GALC in Twitcher Mice. Mol. Ther. 2015, 23, 1681–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricca, A.; Rufo, N.; Ungari, S.; Morena, F.; Martino, S.; Kulik, W.; Alberizzi, V.; Bolino, A.; Bianchi, F.; Del Carro, U.; et al. Combined Gene/Cell Therapies Provide Long-Term and Pervasive Rescue of Multiple Pathological Symptoms in a Murine Model of Globoid Cell Leukodystrophy. Hum. Mol. Genet. 2015, 24, 3372–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, Z.A.; Sanders, D.B.; Massey, J.M. Peripheral neuropathy in Krabbe disease: Effect of Hematopoietic Stem Cell Transplantation. Neurology 2006, 67, 268–272. [Google Scholar] [CrossRef]
- Allewelt, H.; Taskindoust, M.; Troy, J.; Page, K.; Wood, S.; Parikh, S.; Prasad, V.K.; Kurtzberg, J. Long-Term Functional Outcomes after Hematopoietic Stem Cell Transplant for Early Infantile Krabbe Disease. Biol. Blood Marrow Transplant. 2018, 24, 2233–2238. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal Adhesion Regulation of Cell Behavior. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar] [CrossRef]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol 2001, 2, 793–805. [Google Scholar] [CrossRef]
- losegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical Regulation of a Molecular Clutch Defines Force Transmission and Transduction in Response to Matrix Rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-Induced Focal Adhesion Disassembly Is Mediated by Dynamin and Focal Adhesion Kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Ladoux, B.; Mège, R.-M. Mechanobiology of Collective Cell Behaviours. Nat. Rev. Mol. Cell Biol. 2017, 18, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.-H.; Wang, J.-Y.; Chiu, Y.-T.; Wang, M.-P.; Lee, S.-P.; Tai, C.-Y. N-Cadherin Regulates Cell Migration through ARab5-Dependent Temporal Control OfMacropinocytosis. Traffic 2016, 17, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Ribbens, J.; Whiteley, G.; Furuya, H.; Southall, N.; Hu, X.; Marugan, J.; Ferrer, M.; Maegawa, G.H.B. A High-Throughput Screening Assay Using Krabbe Disease Patient Cells. Anal. Biochem. 2013, 434, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Konola, J.T.; Lyerla, T.A.; Skiba, M.C.; Raghavan, S. Establishment of a Galactocerebrosidase-Deficient Twitcher Mouse Cell Line That Expresses Galactocerebrosidase Activity in Hybrids with Control Human Fibroblasts. In Vitro Cell. Dev. Biol. 1988, 24, 575–580. [Google Scholar] [CrossRef]
- Bainbridge, P. Wound Healing and the Role of Fibroblasts. J. Wound Care 2013, 22, 5. [Google Scholar]
- Smith, R.S.; Blieden, T.M.; Phipps, R.P. Fibroblasts as Sentinel Cells. Am. J. Pathol. 1997, 151, 317. [Google Scholar]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 Enables Autophagy-Dependent Focal Adhesion Turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Inui, K.; Tatsumi, N.; Fukushima, H.; Nishigaki, T.; Taniike, M.; Nishimoto, J.; Tsukamoto, H.; Yanagihara, I.; Ozono, K.; et al. Molecular Cloning and Expression of CDNA for Murine Galactocerebrosidase and Mutation Analysis of the Twitcher Mouse, a Model of Krabbe’s Disease. J. Neurochem. 2002, 66, 1118–1124. [Google Scholar] [CrossRef]
- Tonazzini, I.; Jacchetti, E.; Meucci, S.; Beltram, F.; Cecchini, M. Schwann Cell Contact Guidance versus Boundary Interaction in Functional Wound Healing along Nano and Microstructured Membranes. Adv. Healthc. Mater. 2015, 4, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Mezzena, R.; Masciullo, C.; Antonini, S.; Cremisi, F.; Scheffner, M.; Cecchini, M.; Tonazzini, I. Study of Adhesion and Migration Dynamics in Ubiquitin E3A Ligase (UBE3A)-Silenced SYSH5Y Neuroblastoma Cells by Micro-Structured Surfaces. Nanotechnology 2021, 32, 025708. [Google Scholar] [CrossRef] [PubMed]
- Scaccini, L.; Mezzena, R.; De Masi, A.; Gagliardi, M.; Gambarotta, G.; Cecchini, M.; Tonazzini, I. Chitosan Micro-Grooved Membranes with Increased Asymmetry for the Improvement of the Schwann Cell Response in Nerve Regeneration. IJMS 2021, 22, 7901. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hartley, R.; Reiss, B.; Sun, Y.; Pu, J.; Wu, D.; Lin, F.; Hoang, T.; Yamada, S.; Jiang, J.; et al. E-Cadherin Plays an Essential Role in Collective Directional Migration of Large Epithelial Sheets. Cell. Mol. Life Sci. 2012, 69, 2779–2789. [Google Scholar] [CrossRef] [Green Version]
- Tonazzini, I.; Van Woerden, G.M.; Masciullo, C.; Mientjes, E.J.; Elgersma, Y.; Cecchini, M. The Role of Ubiquitin Ligase E3A in Polarized Contact Guidance and Rescue Strategies in UBE3A-Deficient Hippocampal Neurons. Mol. Autism 2019, 10, 41. [Google Scholar] [CrossRef]
- Horzum, U.; Ozdil, B.; Pesen-Okvur, D. Step-by-Step Quantitative Analysis of Focal Adhesions. MethodsX 2014, 1, 56–59. [Google Scholar] [CrossRef]
- Tonazzini, I.; Masciullo, C.; Savi, E.; Sonato, A.; Romanato, F.; Cecchini, M. Neuronal Contact Guidance and YAP Signaling on Ultra-Small Nanogratings. Sci. Rep. 2020, 10, 3742. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- McMurray, R.J.; Dalby, M.J.; Tsimbouri, P.M. Using Biomaterials to Study Stem Cell Mechanotransduction, Growth and Differentiation: Biomaterials for MSCs. J. Tissue Eng. Regen. Med. 2015, 9, 528–539. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, P.; Wang, X.; Pan, P.; Wang, Y.; Zhang, H.; Dong, Y.; Shi, Y.; Jiang, Y.; Yu, R.; et al. YAP Promotes Migration and Invasion of Human Glioma Cells. J. Mol. Neurosci. 2018, 64, 262–272. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of Focal Adhesions and Cancer Cell Migration. Int. J. Cell Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilić, D.; Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Fujimoto, J.; Okada, M.; Yamamoto, T.; et al. Reduced Cell Motility and Enhanced Fochal Adhesion Contact Formation in Cells from FAK-Deficient Mice. Nature 1995, 377, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Angers-Loustau, A.; Côté, J.-F.; Charest, A.; Dowbenko, D.; Spencer, S.; Lasky, L.A.; Tremblay, M.L. Protein Tyrosine Phosphatase-PEST Regulates Focal Adhesion Disassembly, Migration, and Cytokinesis in Fibroblasts. J. Cell Biol. 1999, 144, 1019–1031. [Google Scholar] [CrossRef]
- Mason, D.E.; Collins, J.M.; Dawahare, J.H.; Nguyen, T.D.; Lin, Y.; Voytik-Harbin, S.L.; Zorlutuna, P.; Yoder, M.C.; Boerckel, J.D. YAP and TAZ Limit Cytoskeletal and Focal Adhesion Maturation to Enable Persistent Cell Motility. J. Cell Biol. 2019, 218, 1369–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, W.; Yamada, S. N-Cadherin-Mediated Cell–Cell Adhesion Promotes Cell Migration in a Three-Dimensional Matrix. J. Cell Sci. 2012, 125, 3661–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP Regulates Cell Mechanics by Controlling Focal Adhesion Assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [Green Version]
- Del Grosso, A.; Antonini, S.; Angella, L.; Tonazzini, I.; Signore, G.; Cecchini, M. Lithium Improves Cell Viability in Psychosine-Treated MO3.13 Human Oligodendrocyte Cell Line via Autophagy Activation: Li Improves MO3.13 Cell Viability Via Autophagy Activation. J. Neurosci. Res. 2016, 94, 1246–1260. [Google Scholar] [CrossRef]
- Datla, S.R.; McGrail, D.J.; Vukelic, S.; Huff, L.P.; Lyle, A.N.; Pounkova, L.; Lee, M.; Seidel-Rogol, B.; Khalil, M.K.; Hilenski, L.L.; et al. Poldip2 Controls Vascular Smooth Muscle Cell Migration by Regulating Focal Adhesion Turnover and Force Polarization. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H945–H957. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Kenific, C.M.; Debnath, J. NBR1-Dependent Selective Autophagy Is Required for Efficient Cell-Matrix Adhesion Site Disassembly. Autophagy 2016, 12, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Wittmann, T.; Debnath, J. Autophagy in Adhesion and Migration. J. Cell Sci. 2016, 129, 3685–3693. [Google Scholar] [CrossRef] [Green Version]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of Autophagy with Bafilomycin and Chloroquine Decreases Mitochondrial Quality and Bioenergetic Function in Primary Neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Bhaskar, S. Methods for Detection of Autophagy in Mammalian Cells. In Stem Cells and Aging; Turksen, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 2045, pp. 245–258. ISBN 978-1-4939-9712-1. [Google Scholar]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The Autophagy-Associated Factors DRAM1 and P62 Regulate Cell Migration and Invasion in Glioblastoma Stem Cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-Dependent Production of Secreted Factors Facilitates Oncogenic RAS-Driven Invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock, N.I.; Kreher, C.; Favret, J.; Nguyen, D.; Bongarzone, E.R.; Wrabetz, L.; Feltri, M.L.; Shin, D. Brainstem Development Requires Galactosylceramidase and Is Critical for Pathogenesis in a Model of Krabbe Disease. Nat. Commun. 2020, 11, 5356. [Google Scholar] [CrossRef]
- Valiente, M.; Ciceri, G.; Rico, B.; Marin, O. Focal Adhesion Kinase Modulates Radial Glia-Dependent Neuronal Migration through Connexin-26. J. Neurosci. 2011, 31, 11678–11691. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Rubinsztein, D.C.; Ballabio, A. Lysosomal Storage Diseases as Disorders of Autophagy. Autophagy 2008, 4, 113–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic Ablation of Acid Ceramidase in Krabbe Disease Confirms the Psychosine Hypothesis and Identifies a New Therapeutic Target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103. [Google Scholar] [CrossRef] [Green Version]
- Del Grosso, A.; Parlanti, G.; Angella, L.; Giordano, N.; Tonazzini, I.; Ottalagana, E.; Cecchini, M. Chronic lithium administration in a mouse model for Krabbe disease. JIMD Rep. 2021, 63, 50–65. [Google Scholar] [CrossRef]
- Fuller, M.; Szer, J.; Stark, S.; Fletcher, J.M. Rapid, single-phase extraction of glucosylsphingosine from plasma: A universal screening and monitoring tool. Clin. Chim. Acta 2015, 450, 6–10. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzena, R.; Del Grosso, A.; Pellegrino, R.M.; Alabed, H.B.R.; Emiliani, C.; Tonazzini, I.; Cecchini, M. Mechanotransduction Impairment in Primary Fibroblast Model of Krabbe Disease. Biomedicines 2023, 11, 927. https://doi.org/10.3390/biomedicines11030927
Mezzena R, Del Grosso A, Pellegrino RM, Alabed HBR, Emiliani C, Tonazzini I, Cecchini M. Mechanotransduction Impairment in Primary Fibroblast Model of Krabbe Disease. Biomedicines. 2023; 11(3):927. https://doi.org/10.3390/biomedicines11030927
Chicago/Turabian StyleMezzena, Roberta, Ambra Del Grosso, Roberto Maria Pellegrino, Husam B. R. Alabed, Carla Emiliani, Ilaria Tonazzini, and Marco Cecchini. 2023. "Mechanotransduction Impairment in Primary Fibroblast Model of Krabbe Disease" Biomedicines 11, no. 3: 927. https://doi.org/10.3390/biomedicines11030927
APA StyleMezzena, R., Del Grosso, A., Pellegrino, R. M., Alabed, H. B. R., Emiliani, C., Tonazzini, I., & Cecchini, M. (2023). Mechanotransduction Impairment in Primary Fibroblast Model of Krabbe Disease. Biomedicines, 11(3), 927. https://doi.org/10.3390/biomedicines11030927