

Transcriptional and Epigenetic Alterations in the Progression of Non-Alcoholic Fatty Liver Disease and Biomarkers Helping to Diagnose Non-Alcoholic Steatohepatitis


Abstract
:1. Introduction
2. Materials and Methods
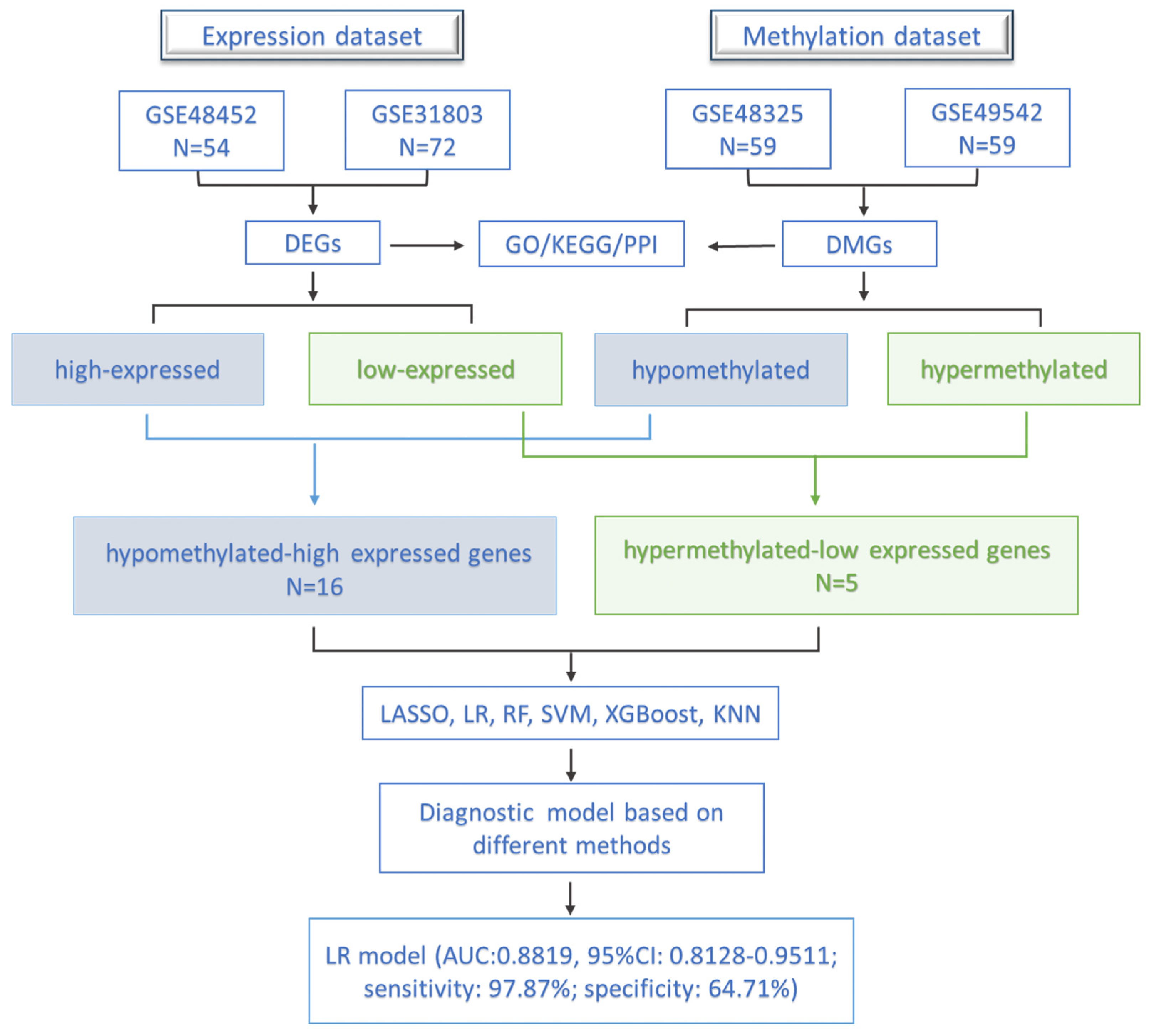
2.1. Data Source and Data Processing
2.2. Identification of DEGs and DMGs in the Progression of NAFLD
2.3. Functional Enrichment Analysis
2.4. Construction and Validation of the Diagnostic Model
2.5. Development and Assessment of the Nomogram
2.6. Validation of the Expression Pattern of the Model Genes in the Testing Cohort
2.7. Development of NAFL and NASH in Mice
2.8. Exploration of the Model Genes Expression Levels in Mice Using qRT-PCR
3. Results
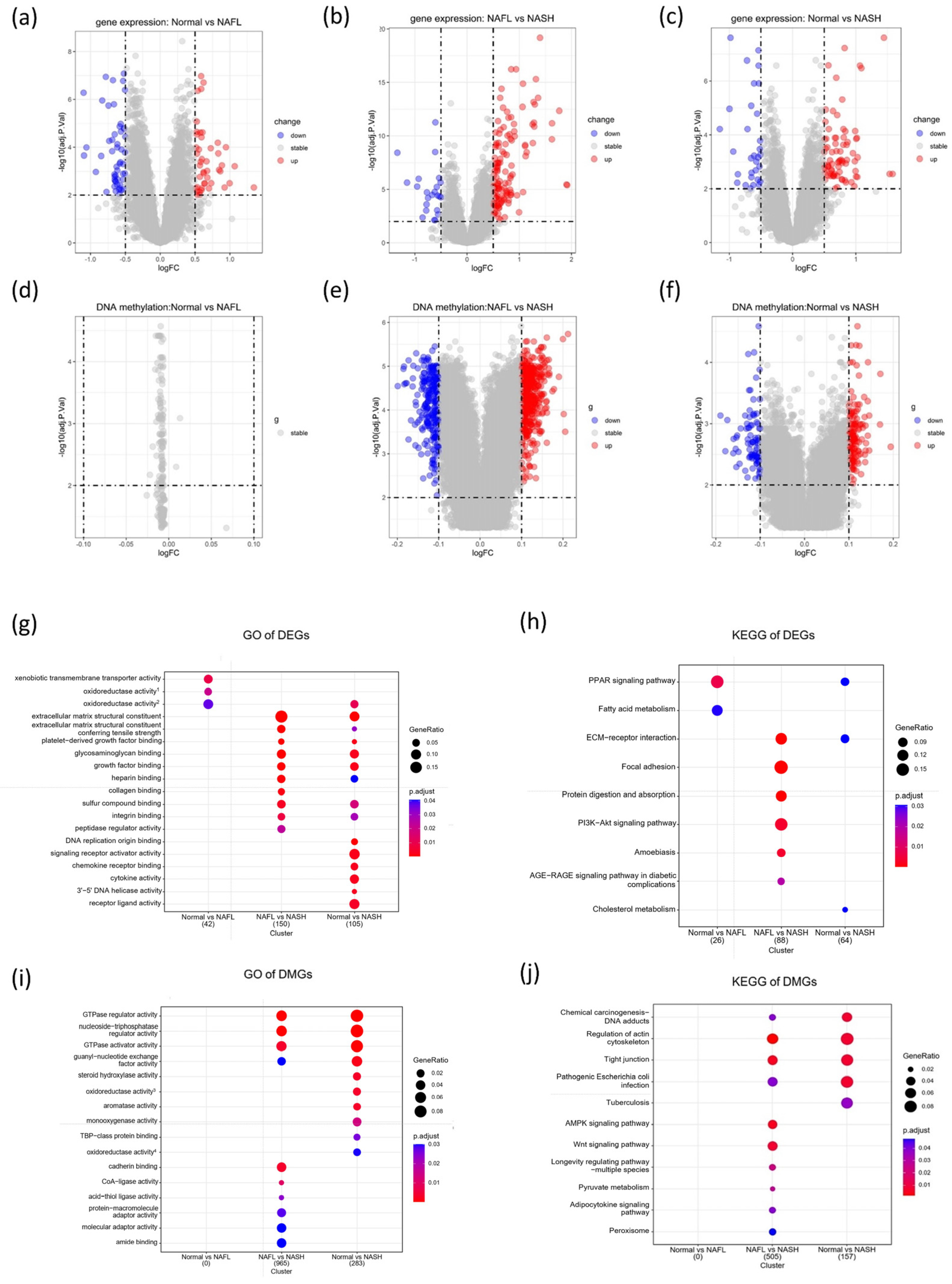
3.1. Identification of DEGs and DMGs in the Progression of NAFLD
3.2. Functional Enrichment Analysis of DEGs
3.3. Functional Enrichment Analysis of DMGs
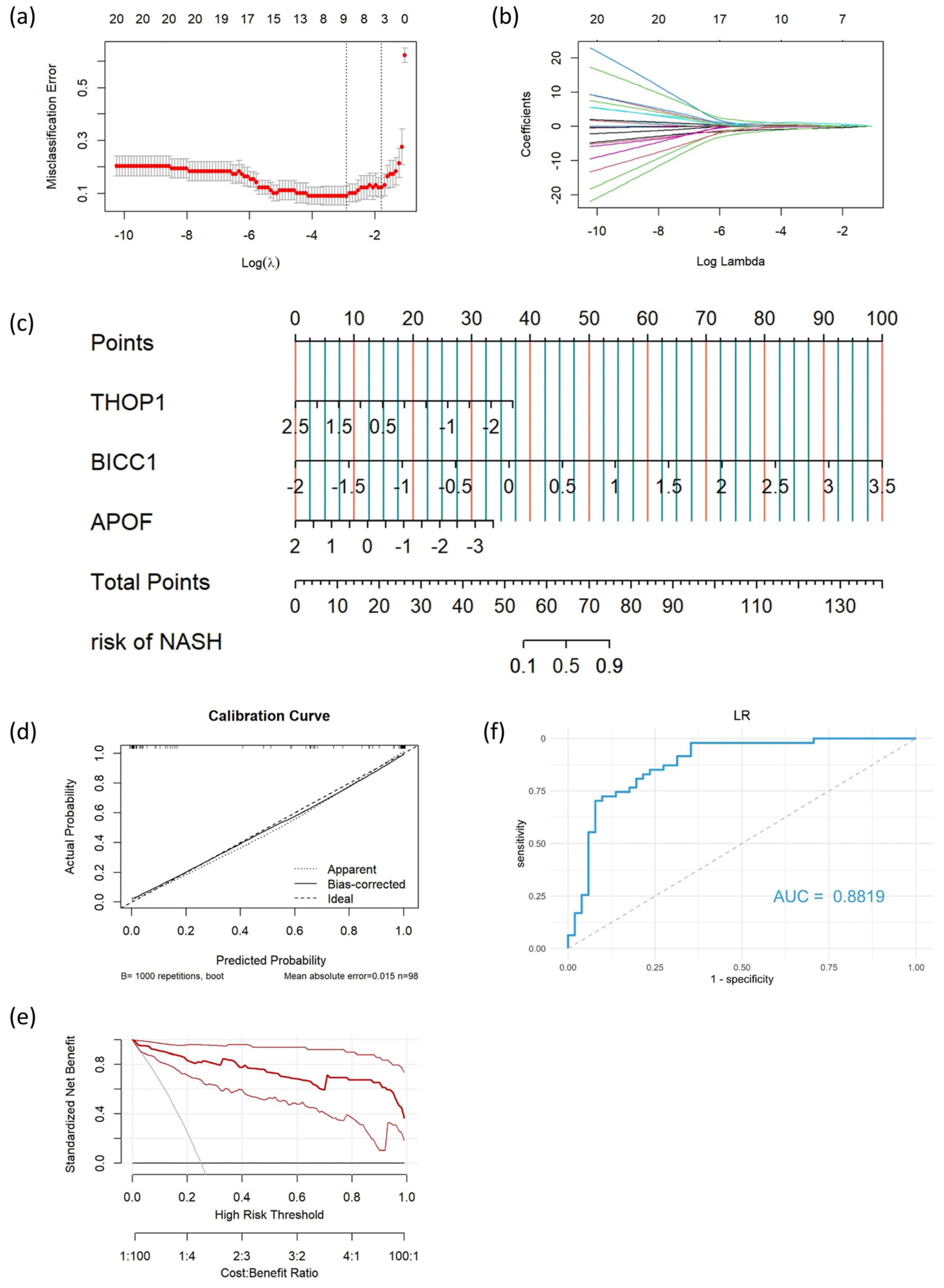
3.4. Construction and Validation of the Diagnostic Model
3.5. Development and Assessment of the Nomogram
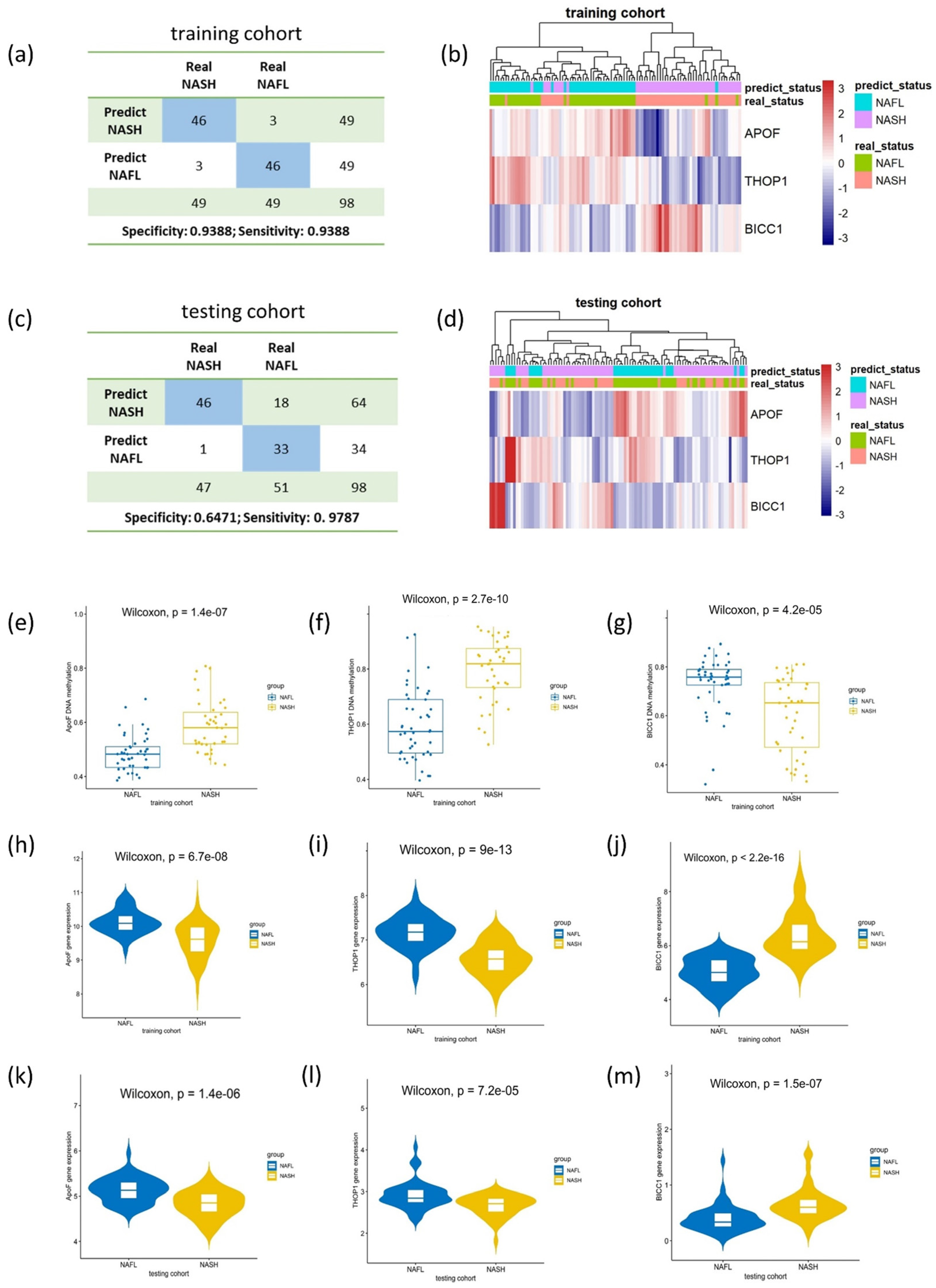
3.6. Validation of the Expression Pattern of the Model Genes in the Testing Cohort
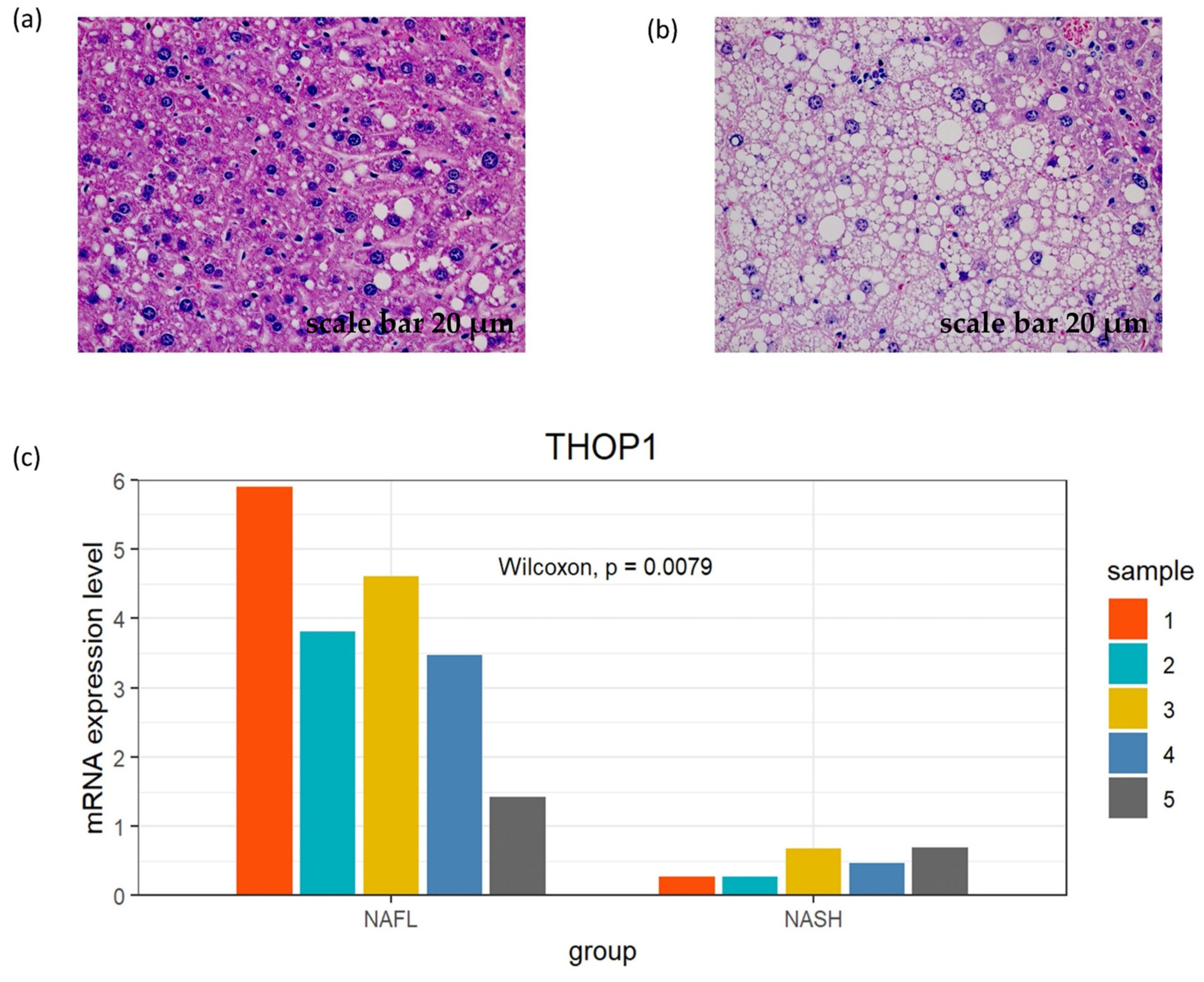
3.7. Exploration of the Expression Pattern of the Model Genes in the Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases from 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; Galiero, R.; Loffredo, G.; Vetrano, E.; Medicamento, G.; Acierno, C.; Rinaldi, L.; Marrone, A.; Salvatore, T.; Monda, M.; et al. Effects of a Combination of Empagliflozin Plus Metformin vs. Metformin Monotherapy on NAFLD Progression in Type 2 Diabetes: The IMAGIN Pilot Study. Biomedicines 2023, 11, 322. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and increased risk of cardiovascular disease: Clinical associations, pathophysiological mechanisms and pharmacological implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus—Mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281.e4. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [Green Version]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Long, J.; Chen, P.; Lin, J.; Bai, Y.; Yang, X.; Bian, J.; Lin, Y.; Wang, D.; Yang, X.; Zheng, Y.; et al. DNA methylation-driven genes for constructing diagnostic, prognostic, and recurrence models for hepatocellular carcinoma. Theranostics 2019, 9, 7251–7267. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef] [PubMed]
- Perakakis, N.; Stefanakis, K.; Mantzoros, C.S. The role of omics in the pathophysiology, diagnosis and treatment of non-alcoholic fatty liver disease. Metabolism 2020, 111, 154320. [Google Scholar] [CrossRef] [PubMed]
- Hendy, O.M.; Rabie, H.; El Fouly, A.; Abdel-Samiee, M.; Abdelmotelb, N.; Elshormilisy, A.A.; Allam, M.; Ali, S.T.; El-Deen, N.M.B.; Abdelsattar, S.; et al. The Circulating Micro-RNAs (-122, -34a and -99a) as Predictive Biomarkers for Non-Alcoholic Fatty Liver Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.N.; Pan, Q.; Zheng, R.D.; Mi, Y.Q.; Shen, F.; Zhou, D.; Chen, G.Y.; Zhu, C.Y.; Fan, J.G. Genome-wide analysis of DNA methylation in human peripheral leukocytes identifies potential biomarkers of nonalcoholic fatty liver disease. Int. J. Mol. Med. 2018, 42, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- He, W.; Huang, C.; Zhang, X.; Wang, D.; Chen, Y.; Zhao, Y.; Li, X. Identification of transcriptomic signatures and crucial pathways involved in non-alcoholic steatohepatitis. Endocrine 2021, 73, 52–64. [Google Scholar] [CrossRef]
- Hernández-Aquino, E.; Muriel, P. Beneficial effects of naringenin in liver diseases: Molecular mechanisms. World J. Gastroenterol. 2018, 24, 1679–1707. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Baumert, T.F. Hepatitis C virus infection and tight junction proteins: The ties that bind. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183296. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut Dysfunction and Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, W.; Xu, J.; Wu, N.; Wang, Y.; Lin, T.; Liu, Y.; Liu, Y.E. coli NF73-1 Isolated From NASH Patients Aggravates NAFLD in Mice by Translocating Into the Liver and Stimulating M1 Polarization. Front. Cell. Infect. Microbiol. 2020, 10, 535940. [Google Scholar] [CrossRef]
- Carvalho, A.T.; Szeler, K.; Vavitsas, K.; Åqvist, J.; Kamerlin, S.C. Modeling the mechanisms of biological GTP hydrolysis. Arch. Biochem. Biophys. 2015, 582, 80–90. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Piazzolla, V.A.; Mangia, A. Noninvasive Diagnosis of NAFLD and NASH. Cells 2020, 9, 1005. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Chang, Z.; Harrison, S.; Lomonaco, R.; Bril, F.; Orsak, B.; Ortiz-Lopez, C.; Hecht, J.; Feldstein, A.E.; Webb, A.; et al. Limited value of plasma cytokeratin-18 as a biomarker for NASH and fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 60, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Xu, D.; Li, Q.; Xie, N.; Xia, J.; Huo, Q.; Li, P.; Chen, Q.; Huang, S. Metabonomics screening of serum identifies pyroglutamate as a diagnostic biomarker for nonalcoholic steatohepatitis. Clin. Chim. Acta 2017, 473, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kozumi, K.; Kodama, T.; Murai, H.; Sakane, S.; Govaere, O.; Cockell, S.; Motooka, D.; Kakita, N.; Yamada, Y.; Kondo, Y.; et al. Transcriptomics Identify Thrombospondin-2 as a Biomarker for NASH and Advanced Liver Fibrosis. Hepatology 2021, 74, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, R.; Shen, F.; Yang, R.; Zhou, D.; Cao, H.; Chen, G.; Pan, Q.; Fan, J. Altered DNA Methylation Sites in Peripheral Blood Leukocytes from Patients with Simple Steatosis and Nonalcoholic Steatohepatitis (NASH). Med. Sci. Monit. 2018, 24, 6946–6967. [Google Scholar] [CrossRef] [PubMed]
- Da, Z.; Gao, L.; Su, G.; Yao, J.; Fu, W.; Zhang, J.; Zhang, X.; Pei, Z.; Yue, P.; Bai, B.; et al. Bioinformatics combined with quantitative proteomics analyses and identification of potential biomarkers in cholangiocarcinoma. Cancer Cell Int. 2020, 20, 130. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.C.; Chen, Y.Z.; Wang, C.H.; Lin, F.J. The nonalcoholic fatty liver disease-like phenotype and lowered serum VLDL are associated with decreased expression and DNA hypermethylation of hepatic ApoB in male offspring of ApoE deficient mothers fed a with Western diet. J. Nutr. Biochem. 2019, 77, 108319. [Google Scholar] [CrossRef]
- Liu, Y.; Morton, R.E. Apolipoprotein F: A natural inhibitor of cholesteryl ester transfer protein and a key regulator of lipoprotein metabolism. Curr. Opin. Lipidol. 2020, 31, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.E.; Mihna, D. Apolipoprotein F concentration, activity, and the properties of LDL controlling ApoF activation in hyperlipidemic plasma. J. Lipid Res. 2022, 63, 100166. [Google Scholar] [CrossRef]
- Seufert, L.; Benzing, T.; Ignarski, M.; Müller, R.U. RNA-binding proteins and their role in kidney disease. Nat. Rev. Nephrol. 2022, 18, 153–170. [Google Scholar] [CrossRef]
- Liu, Y.; Izem, L.; Morton, R.E. Identification of a hormone response element that mediates suppression of APOF by LXR and PPARα agonists. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158583. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gangadharan, B.; Cobbold, J.; Thursz, M.; Zitzmann, N. Absolute quantitation of disease protein biomarkers in a single LC-MS acquisition using apolipoprotein F as an example. Sci. Rep. 2017, 7, 12072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Peng, C.; Song, C.; Zhao, Q.; Rong, J.; Wang, H.; Ding, W.; Wang, F.; Xie, Y. BICC1 as a novel prognostic biomarker in gastric cancer correlating with immune infiltrates. Int. Immunopharmacol. 2020, 87, 106828. [Google Scholar] [CrossRef]
- Wang, H.; Guo, Y.; Mi, N.; Zhou, L. miR-101-3p and miR-199b-5p promote cell apoptosis in oral cancer by targeting BICC1. Mol. Cell. Probes 2020, 52, 101567. [Google Scholar] [CrossRef]
- Orlowski, M.; Michaud, C.; Chu, T.G. A soluble metalloendopeptidase from rat brain. Purification of the enzyme and determination of specificity with synthetic and natural peptides. Eur. J. Biochem. 1983, 135, 81–88. [Google Scholar] [CrossRef]
- Ferro, E.S.; Gewehr, M.C.F.; Navon, A. Thimet Oligopeptidase Biochemical and Biological Significances: Past, Present, and Future Directions. Biomolecules 2020, 10, 1229. [Google Scholar] [CrossRef]
- Gewehr, M.C.F.; Teixeira, A.A.S.; Santos, B.A.C.; Biondo, L.A.; Gozzo, F.C.; Cordibello, A.M.; Eichler, R.A.S.; Reckziegel, P.; Da Silva, R.N.O.; Dos Santos, N.B.; et al. The Relevance of Thimet Oligopeptidase in the Regulation of Energy Metabolism and Diet-Induced Obesity. Biomolecules 2020, 10, 321. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Platform | Mean Age (Years) | Sex (Male/Female) | Viral Hepatitis | Alcohol Use * | Sample Type | Sample Number | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Normal | NAFL | NASH | ||||||||
| Gene expression | GSE48452 | GPL11532 | 46 | 11/43 | 0 | 0 | liver biopsy | 28 | 9 | 17 |
| GSE31803 | GPL570 | N/A | N/A | 0 | 0 | liver biopsy | 0 | 40 | 32 | |
| DNA methylation | GSE48325 | GPL13534 | 48 | 15/44 | 0 | 0 | liver biopsy | 34 | 10 | 15 |
| GSE49542 | GPL13534 | N/A | N/A | 0 | 0 | liver biopsy | 0 | 35 | 24 | |
| Specificity | Sensitivity | PPV | NPV | AUC (95%CI) | ||
|---|---|---|---|---|---|---|
| LR | Training | 93.88% | 93.88% | 93.88% | 93.88% | 0.9792 (0.9575–1.0000) |
| Testing | 64.71% | 97.87% | 71.88% | 97.06% | 0.8819 (0.8128–0.9511) | |
| SVM | Training | 93.88% | 89.80% | 93.62% | 90.20% | 0.9775 (0.9556–0.9994) |
| Testing | 66.67% | 97.87% | 73.02% | 97.14% | 0.8623 (0.7868–0.9378) | |
| KNN | Training | 93.88% | 93.88% | 93.88% | 93.88% | 0.9842 (0.9677–1.0000) |
| Testing | 70.59% | 95.74% | 75.00% | 94.74% | 0.8502 (0.7707–0.9298) | |
| RF | Training | 100.00% | 100.00% | 100.00% | 100.00% | 1.0000 (1.0000–1.0000) |
| Testing | 62.75% | 93.62% | 69.84% | 91.43% | 0.8454 (0.7695–0.9214) | |
| XGBoost | Training | 100.00% | 100.00% | 100.00% | 100.00% | 1.0000 (1.0000–1.0000) |
| Testing | 66.67% | 89.36% | 71.19% | 87.18% | 0.8256 (0.7455–0.9058) | |
| LASSO | Training | 95.92% | 89.80% | 95.65% | 90.38% | 0.9733 (0.9476–0.9991) |
| Testing | 72.55% | 76.60% | 72.00% | 77.08% | 0.8052 (0.7192–0.8911) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Zhang, H.; Jiang, P.; Xie, C.; Luo, Y.; Chen, J. Transcriptional and Epigenetic Alterations in the Progression of Non-Alcoholic Fatty Liver Disease and Biomarkers Helping to Diagnose Non-Alcoholic Steatohepatitis. Biomedicines 2023, 11, 970. https://doi.org/10.3390/biomedicines11030970
Zhu Y, Zhang H, Jiang P, Xie C, Luo Y, Chen J. Transcriptional and Epigenetic Alterations in the Progression of Non-Alcoholic Fatty Liver Disease and Biomarkers Helping to Diagnose Non-Alcoholic Steatohepatitis. Biomedicines. 2023; 11(3):970. https://doi.org/10.3390/biomedicines11030970
Chicago/Turabian StyleZhu, Yalan, He Zhang, Pengjun Jiang, Chengxia Xie, Yao Luo, and Jie Chen. 2023. "Transcriptional and Epigenetic Alterations in the Progression of Non-Alcoholic Fatty Liver Disease and Biomarkers Helping to Diagnose Non-Alcoholic Steatohepatitis" Biomedicines 11, no. 3: 970. https://doi.org/10.3390/biomedicines11030970
APA StyleZhu, Y., Zhang, H., Jiang, P., Xie, C., Luo, Y., & Chen, J. (2023). Transcriptional and Epigenetic Alterations in the Progression of Non-Alcoholic Fatty Liver Disease and Biomarkers Helping to Diagnose Non-Alcoholic Steatohepatitis. Biomedicines, 11(3), 970. https://doi.org/10.3390/biomedicines11030970