
Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance

Abstract
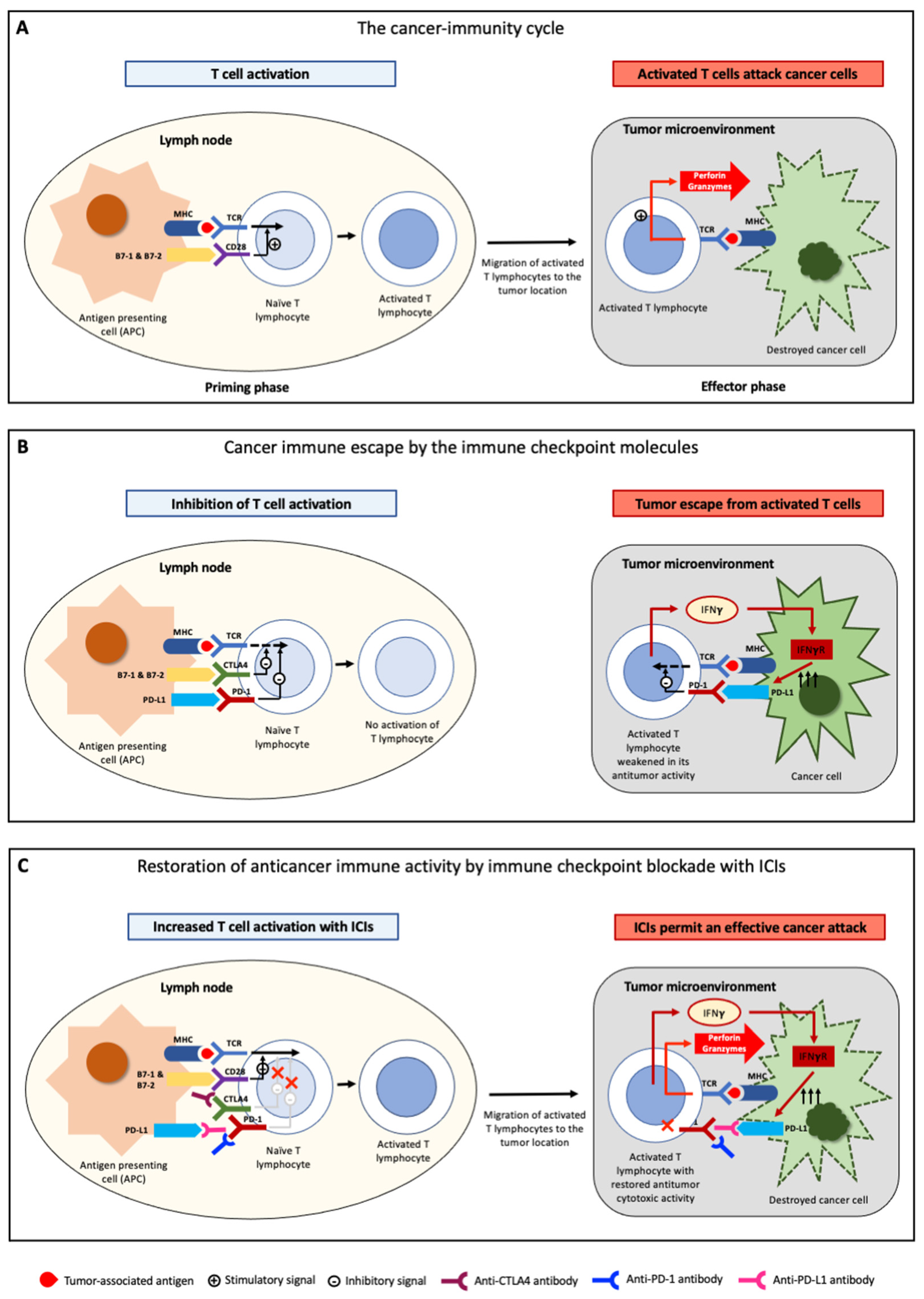
:1. Introduction
2. Antitumor Immunity and Tumor Immune Escape

3. Immune-Checkpoint Inhibitors—Mechanisms of Action
4. Landscape of Immunotherapy for HCC
4.1. Anti-PD1/PD-L1 in Association with Intravenous Anti-VEGF Agents
4.2. Combination of PD-1 and CTLA4 Inhibitors (Dual Checkpoint Blockade)
4.3. Anti-PD1/PD-L1 in Association with TKIs

{kind=link}
| Trial | Phase | Investigational Drug(s) | Comparator | Median OS (HR, 95% CI) | Median PFS (HR, 95% CI) | ORR | Treatment-Related Adverse Event Rates | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Grade ≥ 3 | Most Common (Grade 3–4) | Leading to Discontinuation | Leading to Death | |||||||
| First-line setting | ||||||||||
| IMbrave150 [7,8] | III | Atezolizumab + Bevacizumab (n = 336) | Sorafenib (n = 165) | 19.2 vs. 13.4 months (0.66, 0.52–0.85; p < 0.001) | 6.9 vs. 4.3 months (0.65, 0.53–0.81; p < 0.001) | 30% vs. 11% (p < 0.001) | 43% vs. 46% | Hypertension (12%); AST increase (5%); proteinuria (4%) | 22% vs. 12% | 2% vs. <1% |
| ORIENT-32 [46] | III | Sintilimab + IBI305 (bevacizumab biosimilar) (n = 380) | Sorafenib (n = 191) | NE vs. 10.4 months (0.57, 0.43–0.75; p < 0.0001) | 4.6 vs. 2.8 months (0.56, 0.46–0.70; p < 0.0001) | 21% vs. 4% (p < 0.0001) | 34% vs. 36% | Hypertension (14%); decreased platelet count (8%); proteinuria (5%) | 14% vs. 6% | 2% vs. 1% |
| CheckMate 459 [42] | III | Nivolumab (n = 371) | Sorafenib (n = 372) | 16.4 vs. 14.7 months (0.85, 0.72–1.02; p = 0.075) | 3.7 vs. 3.8 months (0.93, 0.79–1.10; p = ns) | 15% vs. 7% (p = NR) | 22% vs. 49% | AST increase (6%); fatigue (<1%); pruritus (<1%); diarrhea (<1%) | 7% vs. 12% | 1% vs. <1% |
| Cosmic 312 a [49] | III | Atezolizumab + Cabozantinib (n = 432) | Sorafenib (n = 217) | 15.4 vs. 15.5 months (0.90, 0.69–1.18; p = 0.44) | 6.8 vs. 4.2 months (0.63, 0.44–0.91; p = 0.0012) | 11% vs. 4% (p = NR) | 64% vs. 46% | Hypertension (9%); AST increase (9%); ALT increase (9%); PPES (8%) | 14% vs. 8% | 1% vs. <1% |
| HIMALAYA b [9] | III | Tremelimumab + Durvalumab (STRIDE) (n = 393) | Sorafenib (n = 389) | 16.4 vs. 13.8 months (0.78, 0.65–0.93; p = 0.0035) | 3.8 vs. 4.1 months (0.90, 0.75–1.05; p = NR) | 20.1% vs. 5.1% (p = NR) | 25.8% vs. 36.9% | Lipase increase (6.2%); AST increase (5.2%); diarrhea (4.4%); Hyponatremia (4.1%) | 8.25 vs. 11.0% | 2.3% vs. 0.8% |
| Durvalumab (n = 389) | Sorafenib (n = 389) | 16.4 vs. 16.6 months (0.86, 0.73–1.03; noninferiority margin 1.08) | 3.7 vs. 4.1 months (1.02, 0.88–1.19; p = NR) | 17.0% vs. 5.1% (p = NR) | 12.9% vs. 36.9% | AST increase (6.7%); lipase increase (4.1%); ALT increase (3.1%) | 4.1% vs. 11.0% | 0% vs. 0.8% | ||
| LEAP-002 c [50] | III | Lenvatinib + Pembrolizumab (n = 395) | Lenvatinib (n = 399) | 21.2 vs. 19.0 months (0.840, 0.708–0.997; p = 0.0227) | 8.2 vs. 8.0 months (0.867, 0.734–1.024; p = 0.0466) | 26.1% vs. 17.5% (p = NR) | 62.5% vs. 57.5% | NR | NR | 1.0% vs. 0.8% |
| RATIONALE-301 c [52] | III | Tislelizumab (n = 342) | Sorafenib (n = 332) | 15.9 vs. 14.1 months (0.85, 0.71–1.02; p = NR) d | 2.2 vs. 3.5 months (1.10, 0.92–1.33; p = NR) | 14.3% vs. 5.4% (p = NR) | 48.2% vs. 65.4% | NR | 10.9% vs. 18.5% | 4.4% vs. 5.2% |
| NCT03764293 c [51] | III | Camrelizumab + Rivoceranib (Apatinib) (n = 272) | Sorafenib (n = 271) | 22.1 vs. 15.2 months (0.62, 0.49–0.80; p < 0.0001) | 5.6 vs. 3.7 months (0.52, 0.41–0.65; p < 0.0001) | 25.4% vs. 5.9% (p < 0.0001) | 80.9% vs. 52.4% | Hypertension (37.5%); Increased AST (16.5%); increased ALT (12.9%); PPES (12.1%) | 24.3% (3.7% discontinued both drugs) vs. 4.5% | 0.4% vs. 0.4% |
| Second-line setting | ||||||||||
| KEYNOTE-224 [41] | II | Pembrolizumab (n = 104) | - | 12.9 months | 4.9 months | 17% | 26% (irAEs 4%) | Increased AST (7%); increased ALT (4%); fatigue (4%); hyperbilirubinemia (2%) | 23% | 1% |
| KEYNOTE-240 [43] | III | Pembrolizumab (n = 278) | Placebo (n = 135) | 13.9 vs. 10.6 months (0.781, 0.611–0.998; p = 0.0238) | 3.0 vs. 2.8 months (0.718, 0.570–0.904; p = 0.0022) | 18.3% vs. 4.4% (p = 0.00007) | 18.3% vs. 7.5% (irAEs 7.2%) | Increased AST (5.4%); increased ALT (3.6%) | 17.2% vs. 9% | 2.5% vs. 3.0% |
| KEYNOTE-394 c,e [53] | III | Pembrolizumab (n = 300) | Placebo (n = 153) | 14.6 vs. 13.0 months (0.79, 0.63–0.99; p = 0.018) | 2.6 vs. 2.3 months (0.74, 0.60–0.92; p = 0.0032) | 12.7% vs. 1.3% (p = 0.00004) | 14.4% vs. 5.9% | NR | NR | 1% vs. 0% |
| CheckMate 040 [40,54] | I/II | Nivolumab (n = 214 f) | - g | 15.6 months h | 4.0 months i | 14% g | 19% i | Increased AST (4%); increased ALT (2%) | 7% i | 0% |
| CheckMate 040 [47] | I/II | Nivolumab + ipilimumab (n = 50 in arm A j) | - g | 22.8 months | NR | 32% | 53% | Increased AST (16%); increased lipase (12%); increased ALT (8%) | 18% | 2% |
| RESCUE [55] | II | Camrelizumab + Apatinib (n = 120) | - g | NR | 5.5 months | 22.5% | 76.7% (irAEs 7.4%) | Hypertension (34.2%); gGT increase (11.6%); neutropenia (11.1%); increased AST (10.5%); hyperbilirubinemia (10.5%) | 9.2% | 0.8% |
| NCT02519348 [56] | I/II | Durvalumab + Tremelimumab (n = 75 k) | - g | 18.7 months | 2.2 months | 24% | 37.8% (irAEs 12.2%) | Increased AST (12.2%); increased amylase (6.8%); increased lipase (6.8%) | 10.8% | 1.4% |
| NCT01008358 [57] | II | Tremelimumab (n = 21) | - g | 8.2 months | 6.5 months | 17.6% | NR | Increased AST (45%); hyponatremia (30%); increased ALT (25%); encephalopathy (15%) | NR | 0% |
| NCT02989922 [58] | II | Camrelizumab (n = 217) | - g | 13.8 months | 2.1 months | 14.7% | 22% | Increased AST (5%); hyperbilirubinemia (3%); neutropenia (3%) | 4% | 1% |
| RATIONALE-208 [59] | II | Tislelizumab (n = 249) | - g | 13.2 months | 2.7 months | 13% | 15% | Increased AST (3%); increased ALT (1%); hyperbilirubinemia (1%); fatigue (1%) | 5% | 0% |
5. Biomarkers of Response and/or Resistance to Immune Checkpoint Inhibitors
5.1. Markers of Response and/or Resistance in Tumor Genome
5.1.1. Tumor Mutational Burden
5.1.2. Somatic Mutations
5.1.3. Gene Expression Profiling
5.2. Markers of Response and/or Resistance in Tumor Tissue
5.2.1. Immunogenomic Classification in the Prediction of Response to ICIs
5.2.2. PD-L1 Expression
5.2.3. Tumor-Infiltrating Lymphocytes and Other Immune Cells
5.3. Markers of Response and/or Resistance Associated to the Host
5.3.1. Role of Etiology in the Prediction of Response to Immune Checkpoint Inhibitors
5.3.2. The Modulatory Role of Microbiota
5.4. Circulating Markers of Response and/or Resistance
5.5. Clinical Markers of Immune Checkpoint Inhibitors Activity—Immune-Related Adverse Events
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- De Lorenzo, S.; Tovoli, F.; Trevisani, F. Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers 2022, 14, 4616. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Monach, P.A.; Meredith, S.C.; Siegel, C.T.; Schreiber, H. A unique tumor antigen produced by a single amino acid substitution. Immunity 1995, 2, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Dubey, P.; Hendrickson, R.C.; Meredith, S.C.; Siegel, C.T.; Shabanowitz, J.; Skipper, J.C.; Engelhard, V.H.; Hunt, D.F.; Schreiber, H. The immunodominant antigen of an ultraviolet-induced regressor tumor is generated by a somatic point mutation in the DEAD box helicase p68. J. Exp. Med. 1997, 185, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liechtenstein, T.; Dufait, I.; Bricogne, C.; Lanna, A.; Pen, J.; Breckpot, K.; Escors, D. PD-L1/PD-1 Co-Stimulation, a Brake for T cell Activation and a T cell Differentiation Signal. J. Clin. Cell. Immunol. 2012, 12, 006. [Google Scholar] [CrossRef]
- Kudo, M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology 2017, 92 (Suppl. S1), 50–62. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA. 2018, 115, 4749–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maker, A.V.; Attia, P.; Rosenberg, S.A. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J. Immunol. 2005, 175, 7746–7754. [Google Scholar] [CrossRef] [Green Version]
- Agdashian, D.; ElGindi, M.; Xie, C.; Sandhu, M.; Pratt, D.; Kleiner, D.E.; Figg, W.D.; Rytlewski, J.A.; Sanders, C.; Yusko, E.C.; et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Cui, L.; Zhao, X.; Bai, H.; Cai, S.; Wang, G.; Zhao, Z.; Zhao, J.; Chen, S.; Song, J.; et al. Use of Immunotherapy with Programmed Cell Death 1 vs. Programmed Cell Death Ligand 1 Inhibitors in Patients with Cancer: A Systematic Review and Meta-analysis. JAMA Oncol. 2020, 6, 375–384. [Google Scholar] [CrossRef]
- Kurino, T.; Matsuda, R.; Terui, A.; Suzuki, H.; Kokubo, T.; Uehara, T.; Arano, Y.; Hisaka, A.; Hatakeyama, H. Poor outcome with anti-programmed death-ligand 1 (PD-L1) antibody due to poor pharmacokinetic properties in PD-1/PD-L1 blockade-sensitive mouse models. J. Immunother. Cancer 2020, 8, e000400. [Google Scholar] [CrossRef] [Green Version]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Huinen, Z.R.; Huijbers, E.J.M.; van Beijnum, J.R.; Nowak-Sliwinska, P.; Griffioen, A.W. Anti-angiogenic agents—Overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat. Rev. Clin. Oncol. 2021, 18, 527–540. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): A randomised, open-label, phase 2–3 study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Kudo, M. Combination Cancer Immunotherapy in Hepatocellular Carcinoma. Liver Cancer 2018, 7, 20–27. [Google Scholar] [CrossRef]
- Kelley, R.K.; Rimassa, L.; Cheng, A.-L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.-Y.; Ren, Z.; et al. LBA34 Primary results from the phase III LEAP-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first-line (1L) therapy for advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2022, 33, S1401. [Google Scholar] [CrossRef]
- Qin, S.; Chan, L.S.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. LBA35 Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (uHCC): A randomized, phase III trial. Ann. Oncol. 2022, 33, S1401–S1402. [Google Scholar] [CrossRef]
- Qin, S.; Kudo, M.; Meyer, T.; Finn, R.S.; Vogel, A.; Bai, Y.; Guo, Y.; Meng, Z.; Zhang, T.; Satoh, T.; et al. LBA36 Final analysis of RATIONALE-301: Randomized, phase III study of tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Ann. Oncol. 2022, 33, S1402–S1403. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.-Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab plus best supportive care versus placebo plus best supportive care as second-line therapy in patients in Asia with advanced hepatocellular carcinoma (HCC): Phase 3 KEYNOTE-394 study. J. Clin. Oncol. 2022, 40, 383. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Melero, I.; Yau, T.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Choo, S.; Trojan, J.; Welling, T.; Meyer, T.; et al. Impact of antitumor activity on survival outcomes, and nonconventional benefit, with nivolumab (NIVO) in patients with advanced hepatocellular carcinoma (aHCC): Subanalyses of CheckMate-040. J. Clin. Oncol. 2018, 36, 475. [Google Scholar] [CrossRef]
- Xu, J.; Shen, J.; Gu, S.; Zhang, Y.; Wu, L.; Wu, J.; Shao, G.; Zhang, Y.; Xu, L.; Yin, T.; et al. Camrelizumab in Combination with Apatinib in Patients with Advanced Hepatocellular Carcinoma (RESCUE): A Nonrandomized, Open-label, Phase II Trial. Clin. Cancer Res. 2021, 27, 1003–1011. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, D.W.-M.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients with Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef]
- Ren, Z.; Ducreux, M.; Abou-Alfa, G.K.; Merle, P.; Fang, W.; Edeline, J.; Li, Z.; Wu, L.; Assenat, E.; Hu, S.; et al. Tislelizumab in Patients with Previously Treated Advanced Hepatocellular Carcinoma (RATIONALE-208): A Multicenter, Non-Randomized, Open-Label, Phase 2 Trial. Liver Cancer 2022, 12, 72–84. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehí, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Dong, L.-Q.; Peng, L.-H.; Ma, L.-J.; Liu, D.-B.; Zhang, S.; Luo, S.-Z.; Rao, J.-H.; Zhu, H.-W.; Yang, S.-X.; Xi, S.-J.; et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol. 2020, 72, 896–908. [Google Scholar] [CrossRef]
- Bassaganyas, L.; Pinyol, R.; Esteban-Fabró, R.; Torrens, L.; Torrecilla, S.; Willoughby, C.E.; Franch-Expósito, S.; Vila-Casadesús, M.; Salaverria, I.; Montal, R.; et al. Copy-Number Alteration Burden Differentially Impacts Immune Profiles and Molecular Features of Hepatocellular Carcinoma. Clin. Cancer Res. 2020, 26, 6350–6361. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gong, C.; Peng, X.; Bi, X.; Sun, Y.; Zhou, J.; Wu, F.; Zeng, H.; Wang, Y.; Zhou, H.; et al. Serum Concentration of CD137 and Tumor Infiltration by M1 Macrophages Predict the Response to Sintilimab plus Bevacizumab Biosimilar in Advanced Hepatocellular Carcinoma Patients. Clin. Cancer Res. 2022, 28, 3499–3508. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Tan, Z.; Chiu, M.S.; Yang, X.; Yue, M.; Cheung, T.T.; Zhou, D.; Wang, Y.; Chan, A.W.-H.; Yan, C.W.; Kwan, K.Y.; et al. Isoformic PD-1-mediated immunosuppression underlies resistance to PD-1 blockade in hepatocellular carcinoma patients. Gut 2022. [Google Scholar] [CrossRef]
- Zhu, A.X.; Dayyani, F.; Yen, C.-J.; Ren, Z.; Bai, Y.; Meng, Z.; Pan, H.; Dillon, P.; Mhatre, S.K.; Gaillard, V.E.; et al. Alpha-fetoprotein as a potential surrogate biomarker for atezolizumab + bevacizumab treatment of hepatocellular carcinoma. Clin. Cancer Res. 2022, 28, 3537–3545. [Google Scholar] [CrossRef]
- Kim, C.; Yang, H.; Kim, I.; Kang, B.; Kim, H.; Kim, H.; Lee, W.S.; Jung, S.; Lim, H.Y.; Cheon, J.; et al. Association of High Levels of Antidrug Antibodies against Atezolizumab with Clinical Outcomes and T-Cell Responses in Patients with Hepatocellular Carcinoma. JAMA Oncol. 2022, 8, 1825–1829. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Rimini, M.; Rimassa, L.; Ueshima, K.; Burgio, V.; Shigeo, S.; Tada, T.; Suda, G.; Yoo, C.; Cheon, J.; Pinato, D.J.; et al. Atezolizumab plus bevacizumab versus lenvatinib or sorafenib in non-viral unresectable hepatocellular carcinoma: An international propensity score matching analysis. ESMO Open 2022, 7, 100591. [Google Scholar] [CrossRef]
- Pinato, D.J.; Marron, T.U.; Mishra-Kalyani, P.S.; Gong, Y.; Wei, G.; Szafron, D.; Sharon, E.; Saeed, A.; Jun, T.; Dharmapuri, S.; et al. Treatment-related toxicity and improved outcome from immunotherapy in hepatocellular cancer: Evidence from an FDA pooled analysis of landmark clinical trials with validation from routine practice. Eur. J. Cancer 2021, 157, 140–152. [Google Scholar] [CrossRef]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef]
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-Titer Hepatitis B Virus Carrier Mice. Gastroenterology 2020, 158, 1762–1775.e9. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.N.; Fessas, P.; Dominy, K.; Mauri, F.A.; Kaneko, T.; Parcq, P.D.; Khorashad, J.; Toniutto, P.; Goldin, R.D.; Avellini, C.; et al. Qualification of tumour mutational burden by targeted next-generation sequencing as a biomarker in hepatocellular carcinoma. Liver Int. 2021, 41, 192–203. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Haber, P.K.; Castet, F.; Torres-Martin, M.; Andreu-Oller, C.; Puigvehí, M.; Miho, M.; Radu, P.; Dufour, J.-F.; Verslype, C.; Czauderna, C.; et al. Molecular Markers of Response to Anti-PD1 Therapy in Advanced Hepatocellular Carcinoma. Gastroenterology 2023, 164, 72–88. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Poggio, M.; Jin, H.Y.; Shi, Z.; Forester, C.M.; Wang, Y.; Stumpf, C.R.; Xue, L.; Devericks, E.; So, L.; et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019, 25, 301–311. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Zhang, Y.; Cieślik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Investig. 2020, 130, 2712–2726. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.C.N.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018, 67, 931–944. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [Green Version]
- von Felden, J.; Craig, A.J.; Garcia-Lezana, T.; Labgaa, I.; Haber, P.K.; D’Avola, D.; Asgharpour, A.; Dieterich, D.; Bonaccorso, A.; Torres-Martin, M.; et al. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene 2021, 40, 140–151. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef]
- Nsengimana, J.; Laye, J.; Filia, A.; O’Shea, S.; Muralidhar, S.; Poźniak, J.; Droop, A.; Chan, M.; Walker, C.; Parkinson, L.; et al. β-Catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J. Clin. Investig. 2018, 128, 2048–2063. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-P.; Wang, Y.-Q.; Lv, J.-W.; Li, Y.-Q.; Chua, M.L.K.; Le, Q.-T.; Lee, N.; Colevas, A.D.; Seiwert, T.; Hayes, D.N.; et al. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: Implications for immunotherapy. Ann. Oncol. 2019, 30, 68–75. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Pinato, D.J.; Mauri, F.A.; Spina, P.; Cain, O.; Siddique, A.; Goldin, R.; Victor, S.; Pizio, C.; Akarca, A.U.; Boldorini, R.L.; et al. Clinical implications of heterogeneity in PD-L1 immunohistochemical detection in hepatocellular carcinoma: The Blueprint-HCC study. Br. J. Cancer 2019, 120, 1033–1036. [Google Scholar] [CrossRef]
- Zhou, J.; Cheung, A.K.; Liu, H.; Tan, Z.; Tang, X.; Kang, Y.; Du, Y.; Wang, H.; Liu, L.; Chen, Z. Potentiating functional antigen-specific CD8+ T cell immunity by a novel PD1 isoform-based fusion DNA vaccine. Mol. Ther. 2013, 21, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 2022, 71, 2093–2106. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Dizman, N.; Meza, L.; Bergerot, P.; Alcantara, M.; Dorff, T.; Lyou, Y.; Frankel, P.; Cui, Y.; Mira, V.; Llamas, M.; et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: A randomized phase 1 trial. Nat. Med. 2022, 28, 704–712. [Google Scholar] [CrossRef]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for Clinical Benefit of Immune Checkpoint Inhibitor Treatment-A Review from the Melanoma Perspective and Beyond. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Thompson, J.R.; Menon, S.P. Liquid Biopsies and Cancer Immunotherapy. Cancer J. 2018, 24, 78–83. [Google Scholar] [CrossRef]
- Voong, K.R.; Feliciano, J.; Becker, D.; Levy, B. Beyond PD-L1 testing-emerging biomarkers for immunotherapy in non-small cell lung cancer. Ann. Transl. Med. 2017, 5, 376. [Google Scholar] [CrossRef] [Green Version]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef] [Green Version]
- Galle, P.R.; Foerster, F.; Kudo, M.; Chan, S.L.; Llovet, J.M.; Qin, S.; Schelman, W.R.; Chintharlapalli, S.; Abada, P.B.; Sherman, M.; et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 2019, 39, 2214–2229. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Kang, Y.; Yen, C.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations ( REACH-2 ): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Sun, X.; Mei, J.; Lin, W.; Yang, Z.; Peng, W.; Chen, J.; Zhang, Y.; Xu, L.; Chen, M. Reductions in AFP and PIVKA-II can predict the efficiency of anti-PD-1 immunotherapy in HCC patients. BMC Cancer 2021, 21, 775. [Google Scholar] [CrossRef]
- Fessas, P.; Possamai, L.A.; Clark, J.; Daniels, E.; Gudd, C.; Mullish, B.H.; Alexander, J.L.; Pinato, D.J. Immunotoxicity from checkpoint inhibitor therapy: Clinical features and underlying mechanisms. Immunology 2020, 159, 167–177. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Rimola, J.; Díaz-González, Á.; Darnell, A.; Varela, M.; Pons, F.; Hernandez-Guerra, M.; Delgado, M.; Castroagudin, J.; Matilla, A.; Sangro, B.; et al. Complete response under sorafenib in patients with hepatocellular carcinoma: Relationship with dermatologic adverse events. Hepatology 2018, 67, 612–622. [Google Scholar] [CrossRef] [Green Version]
| Response | Refs | Resistance | Ref |
|---|---|---|---|
| Biomarkers of response/resistance in tumor genome | |||
| Activation of IFN signaling pathways 4-genes signature 11-genes signature | [60,61] | Copy number alterations (CNA)—loss of heterozygosity of HLA alleles CTNNB1 mutations Activation of WNT/β-catenin pathway | [34,62,63] [64] [64,65] |
| Biomarkers of response/resistance in tumor tissue | |||
| ↑ T cell infiltration M1 macrophage polarization ↑ cytotoxicity INFγ, PRF1, GZMB, TBX21, KLRK1 ↑ Immune cell homing CXCL9, CXCL10 Antigen presentation—dendritic cells PD-L1 expression in cancer cells and/or immune cells | [34,66] [8,33,34] [34] [34,61] [34] [35,40,41,61] | ↑ cancer-associated fibroblast ↑ stromal activation M2 macrophage polarization ↑ CD8+ PD1+ T cells CCL5, CCL3, GZMK, GZMA, CXCR6 ↑ CD4 Treg cells ↑ Δ42PD-1+ tumor-infiltrating T cells | [34] [33,34] [33,34] [67] [34] [68] |
| Biomarkers of response/resistance in blood | |||
| ↑ Baseline CD137 ↓ AFP during treatment | [66] [69] | Antidrug antibodies (ADAs) | [3,70] |
| Biomarkers of response/resistance associated to the host | |||
| Gut microbiota diversity | [71] | Etiology—NASH | [60,67,72] |
| Clinical markers of immunotherapy activity | |||
| Development of immune-related adverse events | [73] | ||
| Inflamed Class (~35%) | Non-Inflamed Class (~65%) | |||||
|---|---|---|---|---|---|---|
| Active | Exhausted | Immune-Like | Intermediate | Excluded | ||
| Tumor immune microenvironment | Immune infiltrate | |||||
| + + + + | + + + − | + + + − | + − − − | − − − − | ||
| ↑ TIL/TLS abundance | ↓ TIL/TLS abundance | |||||
| ↑ CD8+ T cells and M1 macrophages | ↑ M2 macrophages and exhausted T cells | ↑ CD8+ T cells and M1 macrophages | ↑ M2 macrophages and Treg cells | |||
| ↑ T cells/nucleated cells fraction, polyclonal T cell expansion and diverse TCR repertoire | Oligoclonal expansion | ↓ T cells/nucleated cells fraction and diverse TCR repertoire | ||||
| ↑ immune-checkpoint molecules/CD8 | ↓ immune-checkpoint molecules/CD8 | |||||
| ↑ PD-L1 | ↑ PD-L1 | ↑ LAG3 ↑ CTLA4 | ↓ LAG3 ↓ CTLA4 | ↓ CD8 ↓ PD-L1 | ||
| ↑ IFNγ, GZMB, PRF1, PD-1 signaling | ↓ IFNγ, GZMB, PRF1, PD-1 signaling | |||||
| Liquid Biopsy-based cytokine prediction of the inflamed class | ↓ CXCL9, CXCL10, CXCL11, CCL5 | |||||
| Molecular characterization | ↑ TGFβ | ↑ Wnt-β catenin | ↑ Wnt-β catenin | ↑ PTK2 | ||
| ↑ Hoshida S1 | ↑ Hoshida S2 | ↑ Hoshida S2/3 | ||||
| ↑ Chiang IFN | ↑ Chiang CTNNB1 | ↑ Chiang Poly7 | ↑ Chiang CTNNB1 | |||
| Genomic characterization | Chromosomal aberrations | |||||
| − − − − | + − − − | + + − − | + + + − | + + + + | ||
| ↑ deletions at 16p13.13, 4q21.1, 4q35.1 | Amplification 8q24.3 | |||||
| ↑ CTNNB1 mutation | ↑ TP53 mutation | ↑ CTNNB1 mutation | ||||
| Epigenetic features | 192 immune-related genes differentially methylated | Hypomethylation of HLA-I related genes | Hypomethylation of PTK2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelizzaro, F.; Farinati, F.; Trevisani, F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines 2023, 11, 1020. https://doi.org/10.3390/biomedicines11041020
Pelizzaro F, Farinati F, Trevisani F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines. 2023; 11(4):1020. https://doi.org/10.3390/biomedicines11041020
Chicago/Turabian StylePelizzaro, Filippo, Fabio Farinati, and Franco Trevisani. 2023. "Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance" Biomedicines 11, no. 4: 1020. https://doi.org/10.3390/biomedicines11041020
APA StylePelizzaro, F., Farinati, F., & Trevisani, F. (2023). Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines, 11(4), 1020. https://doi.org/10.3390/biomedicines11041020