
Dimethyl Fumarate as Potential Treatment for Alzheimer’s Disease: Rationale and Clinical Trial Design

Abstract
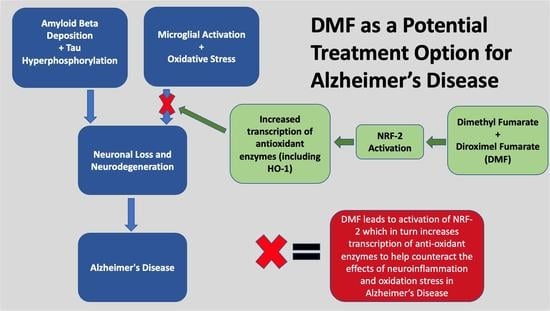
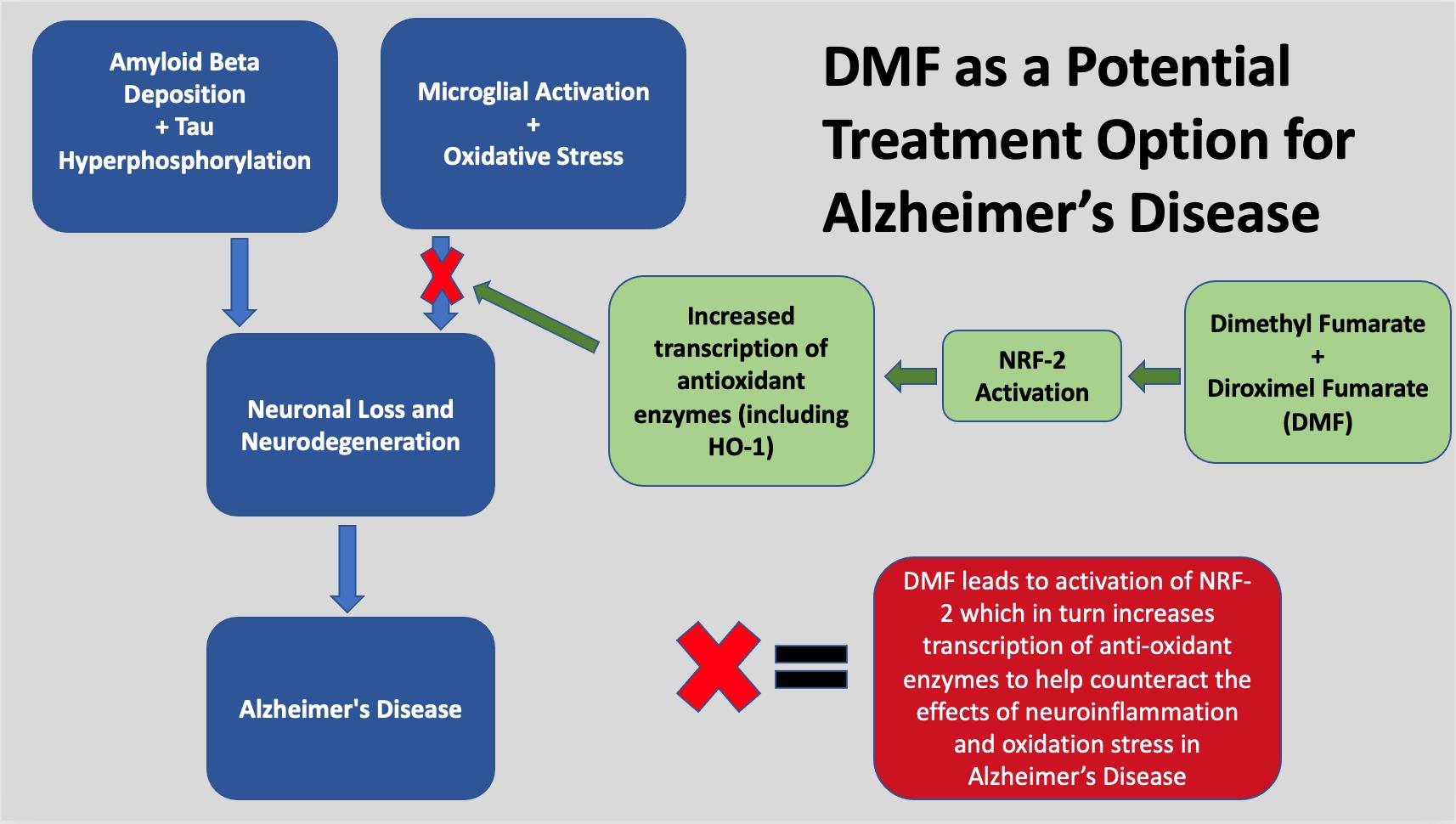
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Overview of Alzheimer’s Disease
2. Materials and Methods
3. Microglial Activation and AD
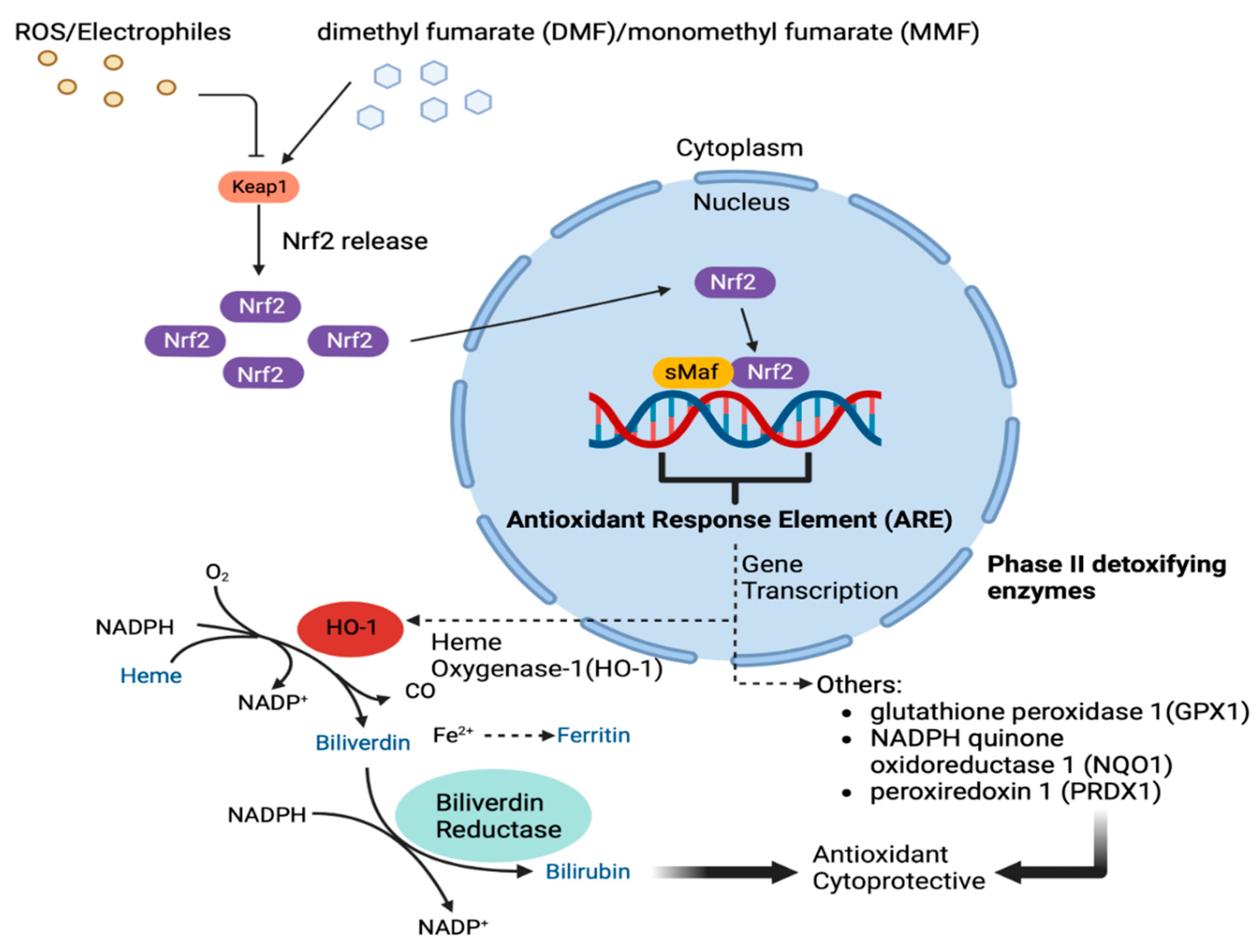
4. The Role of NRF-2
5. The Role of Heme Oxygenase
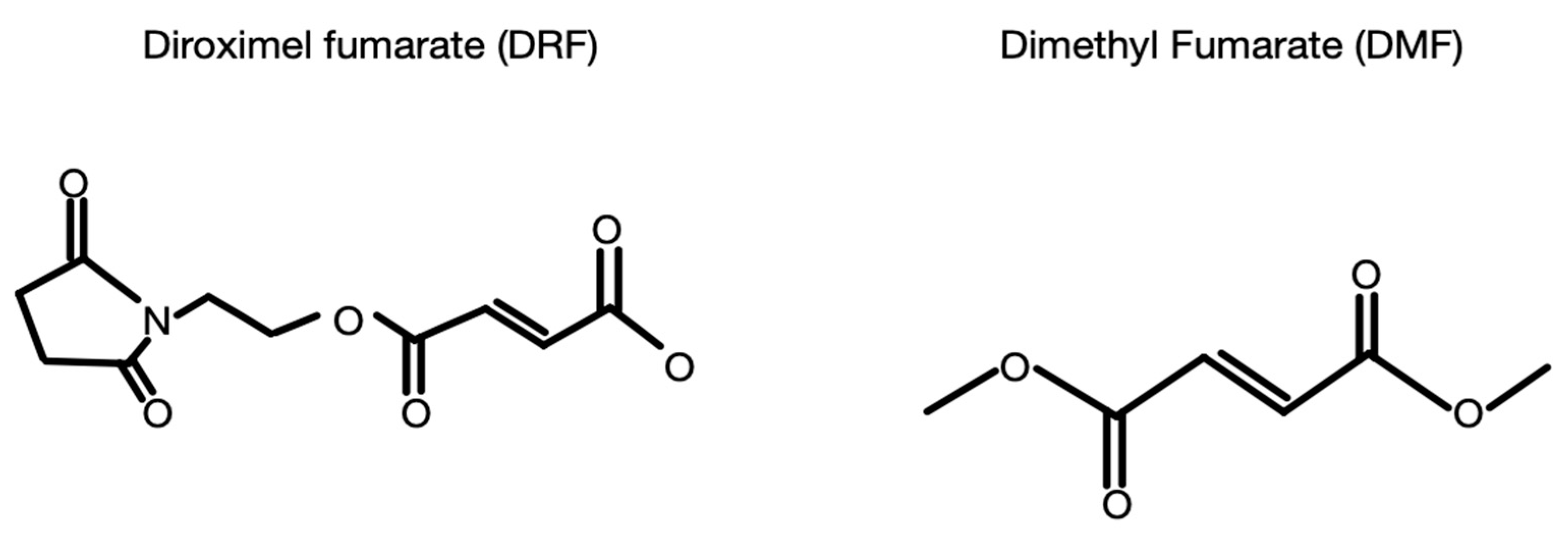
6. Dimethyl Fumarate and Diroximel Fumarate
7. Rationale for a Clinical Trial of Dimethyl Fumarate/Diroximel Fumarate in Mild Cognitive Impairment and AD
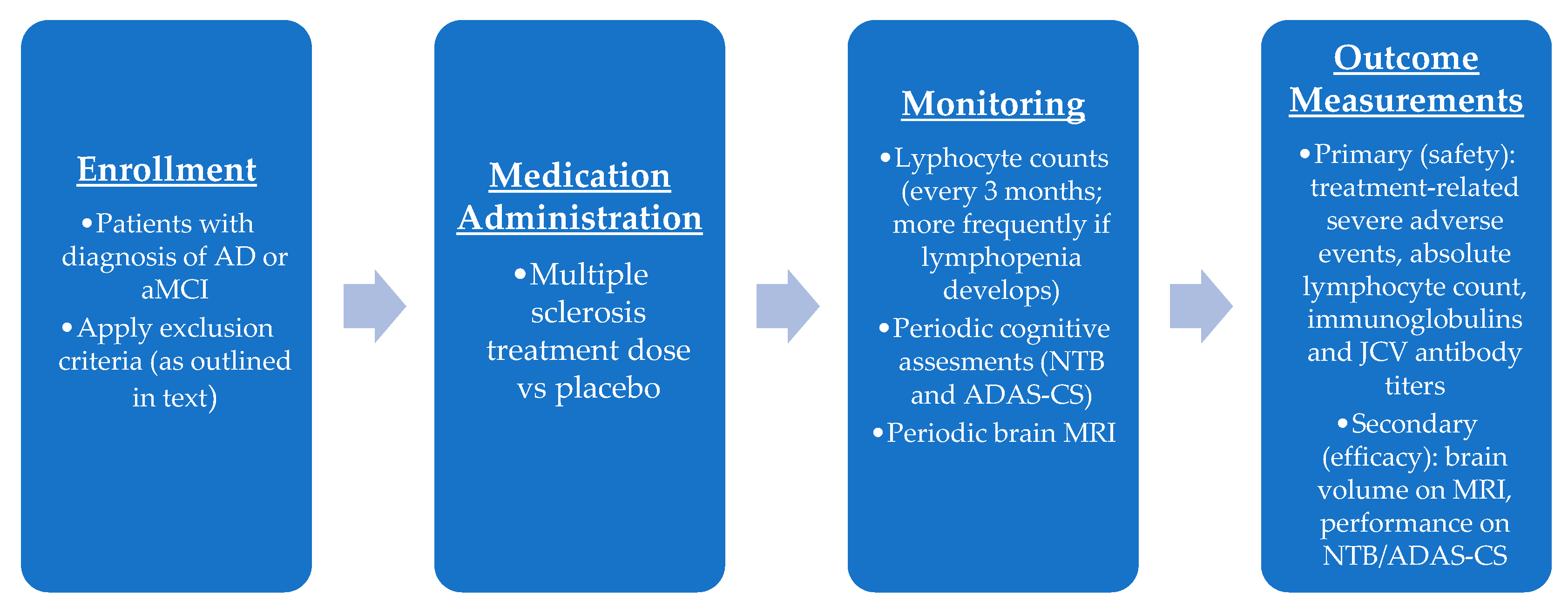
7.1. Phase I Trial
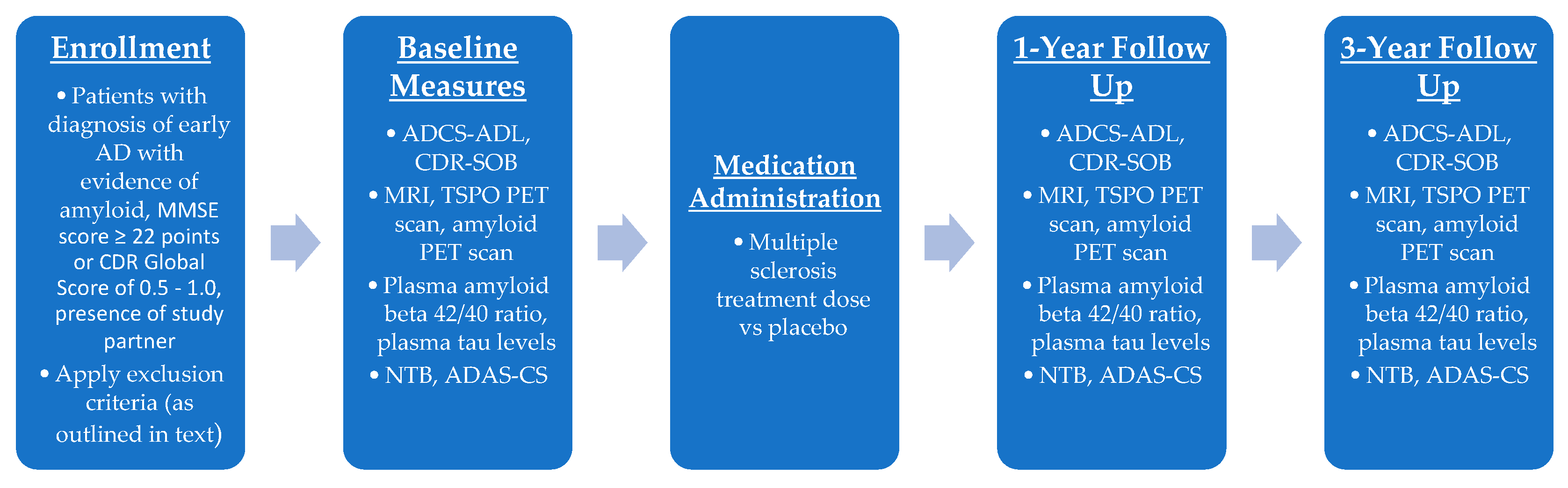
7.2. Phase II Trial
7.3. Future Considerations
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Piazza, F.; Caminiti, S.P.; Zedde, M.; Presotto, L.; DiFrancesco, J.C.; Pascarella, R.; Giossi, A.; Sessa, M.; Poli, L.; Basso, G.; et al. Association of Microglial Activation with Spontaneous ARIA-E and CSF Levels of Anti-Aβ Autoantibodies. Neurology 2022, 99, e1265–e1277. [Google Scholar] [CrossRef]
- Adhikari, U.K.; Adhikari, U.K.; Khan, R.; Mikhael, M.; Balez, R.; David, M.A.; Mahns, D.; Hardy, J.; Tayebi, M. Therapeutic anti-amyloid β antibodies cause neuronal disturbances. Alzheimer’s Dement. 2022. early view. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, E.; Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nuñez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J. Vitagenes, cellular stress response, and acetylcarnitine: Relevance to hormesis. Biofactors 2009, 35, 146–160. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Scuto, M.; Fusco, R.; Trovato, A.; Ontario, M.L.; Crea, R.; Di Paola, R.; Cuzzocrea, S.; Calabrese, V. Anti-inflammatory and Anti-oxidant Activity of Hidrox(®) in Rotenone-Induced Parkinson’s Disease in Mice. Antioxidants 2020, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zheng, Y.; Huang, L.; Ye, J.; Ye, Y.; Luo, H.; Chen, X.; Yao, W.; Chen, J.; Zhang, J.C. Nrf2 regulates the arginase 1+ microglia phenotype through the initiation of TREM2 transcription, ameliorating depression-like behavior in mice. Transl. Psychiatry 2022, 12, 459. [Google Scholar] [CrossRef]
- Doré, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [CrossRef]
- Chang, E.F.; Wong, R.J.; Vreman, H.J.; Igarashi, T.; Galo, E.; Sharp, F.R.; Stevenson, D.K.; Noble-Haeusslein, L.J. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J. Neurosci. 2003, 23, 3689–3696. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, H.; Doré, S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol. Dis. 2006, 22, 473–476. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W. A heme oxygenase-1 transducer model of degenerative and developmental brain disorders. Int. J. Mol. Sci. 2015, 16, 5400–5419. [Google Scholar] [CrossRef]
- Parfenova, H.; Basuroy, S.; Bhattacharya, S.; Tcheranova, D.; Qu, Y.; Regan, R.F.; Leffler, C.W. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: Contributions of HO-1 and HO-2 to cytoprotection. Am. J. Physiol. Cell Physiol. 2006, 290, C1399–C1410. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Cissé, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Mehindate, K.; Sahlas, D.J.; Frankel, D.; Mawal, Y.; Liberman, A.; Corcos, J.; Dion, S.; Schipper, H.M. Proinflammatory cytokines promote glial heme oxygenase-1 expression and mitochondrial iron deposition: Implications for multiple sclerosis. J. Neurochem. 2001, 77, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase-1 in Alzheimer disease: A tribute to Moussa Youdim. J. Neural Transm. 2011, 118, 381–387. [Google Scholar] [CrossRef]
- Jayanti, S.; Vítek, L.; Tiribelli, C.; Gazzin, S. The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases. Antioxidants 2020, 9, 900. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β1-42-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef]
- Takeda, A.; Perry, G.; Abraham, N.G.; Dwyer, B.E.; Kutty, R.K.; Laitinen, J.T.; Petersen, R.B.; Smith, M.A. Overexpression of heme oxygenase in neuronal cells, the possible interaction with Tau. J. Biol. Chem. 2000, 275, 5395–5399. [Google Scholar] [CrossRef]
- Jiao, W.; Wang, Y.; Kong, L.; Ou-Yang, T.; Meng, Q.; Fu, Q.; Hu, Z. CART peptide activates the Nrf2/HO-1 antioxidant pathway and protects hippocampal neurons in a rat model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 501, 1016–1022. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Rosito, M.; Testi, C.; Parisi, G.; Cortese, B.; Baiocco, P.; Di Angelantonio, S. Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment. Antioxidants 2020, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 2011, 6, e16172. [Google Scholar] [CrossRef] [PubMed]
- Casili, G.; Cordaro, M.; Impellizzeri, D.; Bruschetta, G.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Reduces Inflammatory Responses in Experimental Colitis. J. Crohn’s Colitis 2016, 10, 472–483. [Google Scholar] [CrossRef]
- Demir, S.; Heckers, S.; Pedreiturria, X.; Hess, D.; Trampe, A.K.; Chan, A.; Gold, R. Low dose fumaric acid esters are effective in a mouse model of spontaneous chronic encephalomyelitis. J. Neuroimmunol. 2015, 285, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewska, A.P.; Seta, F.; Han, R.; Czajka, C.A.; Makino, K.; Stawski, L.; Isenberg, J.S.; Browning, J.L.; Trojanowska, M. Dimethyl Fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci. Rep. 2017, 7, 41605. [Google Scholar] [CrossRef] [PubMed]
- Seidel, P.; Roth, M. Anti-inflammatory dimethylfumarate: A potential new therapy for asthma? Mediat. Inflamm. 2013, 2013, 875403. [Google Scholar] [CrossRef]
- Safari, A.; Khodabandeh, Z.; Borhani-Haghighi, A. Dimethyl Fumarate Can Enhance the Potential Therapeutic Effects of Epidermal Neural Crest Stem Cells in COVID-19 Patients. Stem Cell Rev. Rep. 2021, 17, 300–301. [Google Scholar] [CrossRef]
- Timpani, C.A.; Rybalka, E. Calming the (Cytokine) Storm: Dimethyl Fumarate as a Therapeutic Candidate for COVID-19. Pharmaceuticals 2020, 14, 15. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938, Correction in Nat. Commun. 2020, 11, 5419. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Grebenchikov, O.A. Transcription Factor Nrf2 as a Potential Therapeutic Target for Prevention of Cytokine Storm in COVID-19 Patients. Biochemistry 2020, 85, 833–837. [Google Scholar] [CrossRef]
- Al-Ani, M.; Elemam, N.M.; Hundt, J.E.; Maghazachi, A.A. Drugs for Multiple Sclerosis Activate Natural Killer Cells: Do They Protect Against COVID-19 Infection? Infect. Drug Resist. 2020, 13, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Omarjee, L.; Perrot, F.; Meilhac, O.; Mahe, G.; Bousquet, G.; Janin, A. Immunometabolism at the cornerstone of inflammaging, immunosenescence, and autoimmunity in COVID-19. Aging 2020, 12, 26263–26278. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Blatnik, M.; Thorpe, S.R.; Baynes, J.W. Succination of proteins by fumarate: Mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008, 1126, 272–275. [Google Scholar] [CrossRef]
- Frizzell, N.; Rajesh, M.; Jepson, M.J.; Nagai, R.; Carson, J.A.; Thorpe, S.R.; Baynes, J.W. Succination of thiol groups in adipose tissue proteins in diabetes: Succination inhibits polymerization and secretion of adiponectin. J. Biol. Chem. 2009, 284, 25772–25781. [Google Scholar] [CrossRef]
- Humphries, F.; Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Manuel, A.M.; Frizzell, N. Adipocyte protein modification by Krebs cycle intermediates and fumarate ester-derived succination. Amino Acids 2013, 45, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Merkley, E.D.; Metz, T.O.; Smith, R.D.; Baynes, J.W.; Frizzell, N. The succinated proteome. Mass Spectrom. Rev. 2014, 33, 98–109. [Google Scholar] [CrossRef]
- Dickens, A.M.; Dickens, A.M.; Anthony, D.C.; Deutsch, R.; Mielke, M.M.; Claridge, T.D.; Grant, I.; Franklin, D.; Rosario, D.; Marcotte, T.; et al. Cerebrospinal fluid metabolomics implicate bioenergetic adaptation as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. Aids 2015, 29, 559–569. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134 Pt 3, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Landeck, L.; Asadullah, K.; Amasuno, A.; Pau-Charles, I.; Mrowietz, U. Dimethyl fumarate (DMF) vs. monoethyl fumarate (MEF) salts for the treatment of plaque psoriasis: A review of clinical data. Arch. Dermatol. Res. 2018, 310, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Fox, R.J.; Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Xu, H.N.; Vance, P.; Gruenewald, A.L.; Garza, R.; Midkiff, C.; Alvarez-Hernandez, X.; Irwin, D.J.; Gill, A.J.; Kolson, D.L. Dimethyl fumarate, an approved multiple sclerosis treatment, reduces brain oxidative stress in SIV-infected rhesus macaques: Potential therapeutic repurposing for HIV neuroprotection. Antioxidants 2021, 10, 416. [Google Scholar] [CrossRef]
- Sun, X.; Suo, X.; Xia, X.; Yu, C.; Dou, Y. Dimethyl Fumarate is a Potential Therapeutic Option for Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 443–456. [Google Scholar] [CrossRef]
- Jordan, A.L.; Yang, J.; Fisher, C.J.; Racke, M.K.; Mao-Draayer, Y. Progressive multifocal leukoencephalopathy in dimethyl fumarate-treated multiple sclerosis patients. Mult. Scler. J. 2022, 28, 7–15. [Google Scholar] [CrossRef]
- Naismith, R.T.; Naismith, R.T.; Wolinsky, J.S.; Wundes, A.; LaGanke, C.; Arnold, D.L.; Obradovic, D.; Freedman, M.S.; Gudesblatt, M.; Ziemssen, T.; et al. Diroximel fumarate (DRF) in patients with relapsing-remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study. Mult. Scler. 2020, 26, 1729–1739. [Google Scholar] [CrossRef]
- Amato, M.P.; Goretti, B.; Brescia Morra, V.; Gallo, P.; Zaffaroni, M.; Onofrj, M.; Cocco, E.; Borriello, G.; Zipoli, V.; Trojano, M. Effects of 2-year treatment with dimethyl fumarate on cognition and functional impairment in patients with relapsing remitting multiple sclerosis. Neurol. Sci. 2020, 41, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Plucińska, K.; Jerzemowska, G.; Wrona, D. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav. Brain Res. 2016, 308, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Fatah, I.M.; Abdelrazek, H.M.A.; Ibrahim, S.M.; Abdallah, D.M.; El-Abhar, H.S. Dimethyl fumarate abridged tauo-/amyloidopathy in a D-Galactose/ovariectomy-induced Alzheimer’s-like disease: Modulation of AMPK/SIRT-1, AKT/CREB/BDNF, AKT/GSK-3β, adiponectin/Adipo1R, and NF-κB/IL-1β/ROS trajectories. Neurochem. Int. 2021, 148, 105082, Correction in Neurochem. Int. 2023, 11, 105523. [Google Scholar] [CrossRef] [PubMed]
- A Phase 2 Study to Evaluate Efficacy and Safety of AL002 in Participants with Early Alzheimer’s Disease (INVOKE-2). Registry Name Identifier: NCT04592874. Available online: https://clinicaltrials.gov/ct2/show/NCT04592874?term=INVOKE-2&draw=2&rank=1 (accessed on 4 May 2023).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharkus, R.; Thakkar, R.; Kolson, D.L.; Constantinescu, C.S. Dimethyl Fumarate as Potential Treatment for Alzheimer’s Disease: Rationale and Clinical Trial Design. Biomedicines 2023, 11, 1387. https://doi.org/10.3390/biomedicines11051387
Sharkus R, Thakkar R, Kolson DL, Constantinescu CS. Dimethyl Fumarate as Potential Treatment for Alzheimer’s Disease: Rationale and Clinical Trial Design. Biomedicines. 2023; 11(5):1387. https://doi.org/10.3390/biomedicines11051387
Chicago/Turabian StyleSharkus, Robert, Richa Thakkar, Dennis L. Kolson, and Cris S. Constantinescu. 2023. "Dimethyl Fumarate as Potential Treatment for Alzheimer’s Disease: Rationale and Clinical Trial Design" Biomedicines 11, no. 5: 1387. https://doi.org/10.3390/biomedicines11051387
APA StyleSharkus, R., Thakkar, R., Kolson, D. L., & Constantinescu, C. S. (2023). Dimethyl Fumarate as Potential Treatment for Alzheimer’s Disease: Rationale and Clinical Trial Design. Biomedicines, 11(5), 1387. https://doi.org/10.3390/biomedicines11051387