
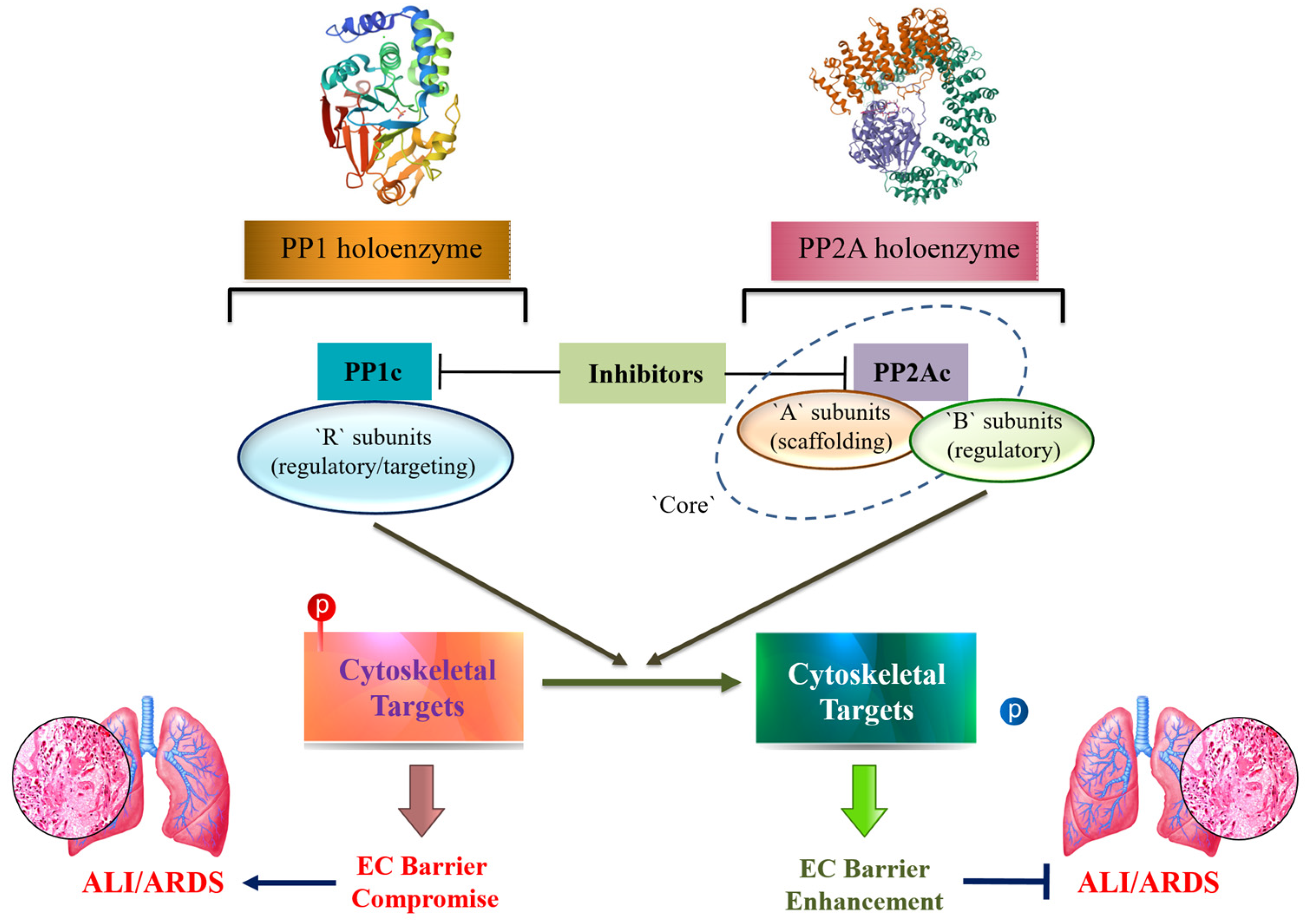
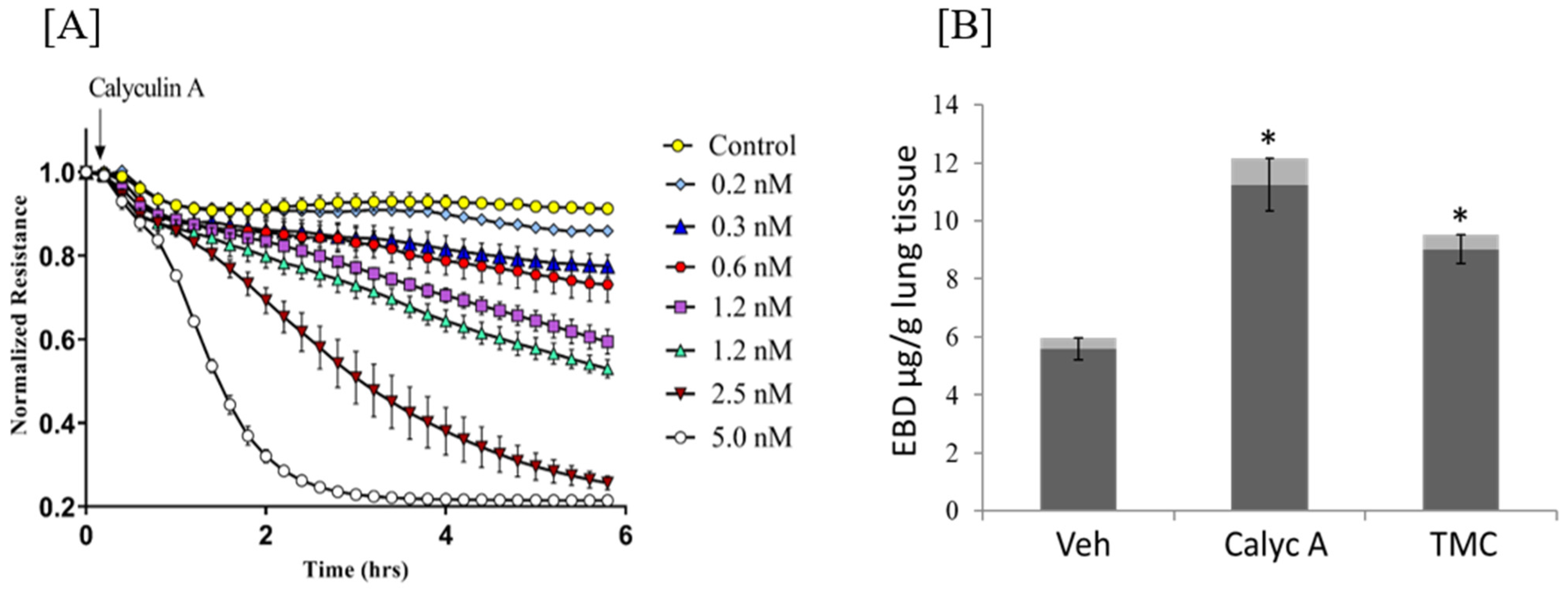
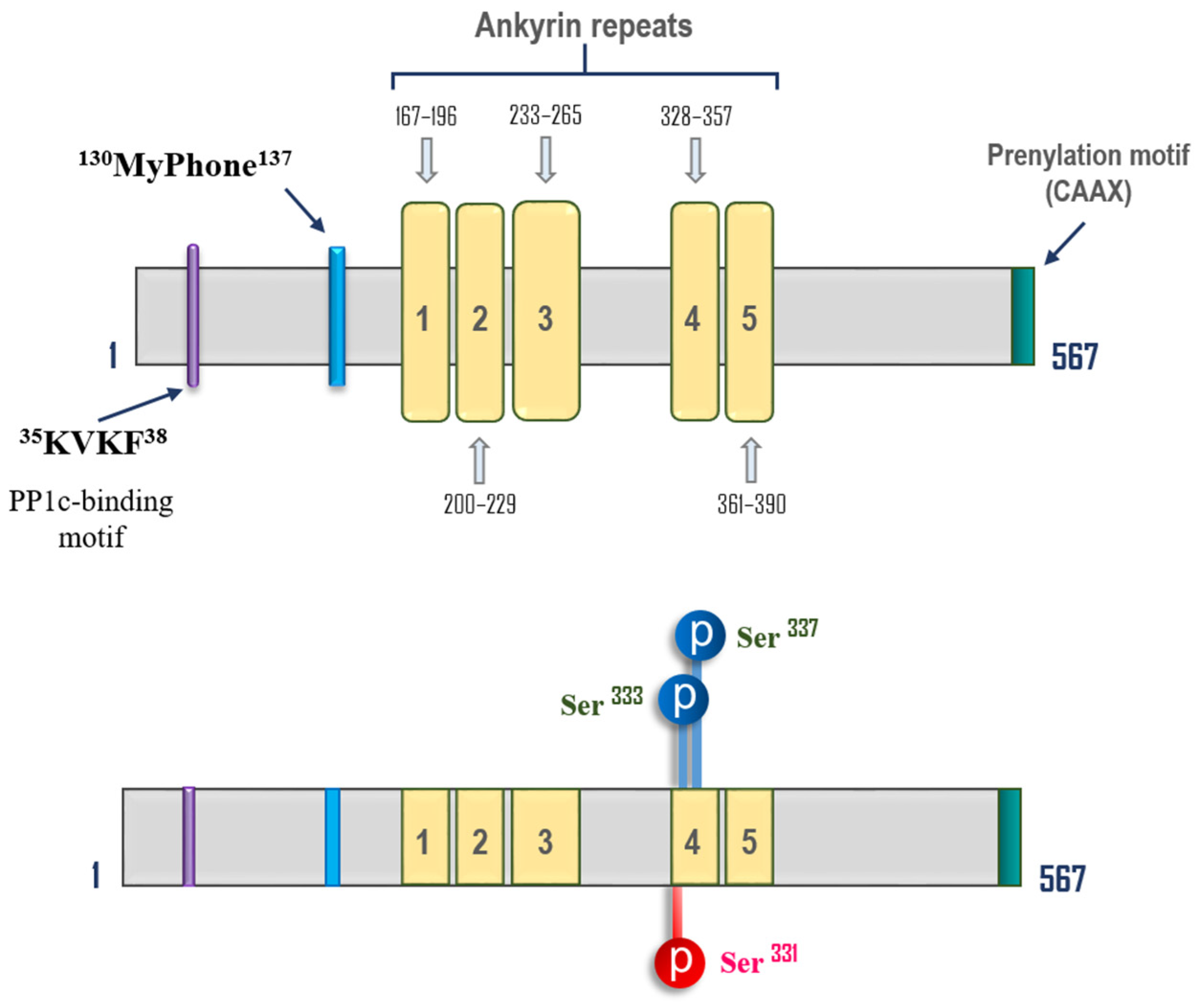
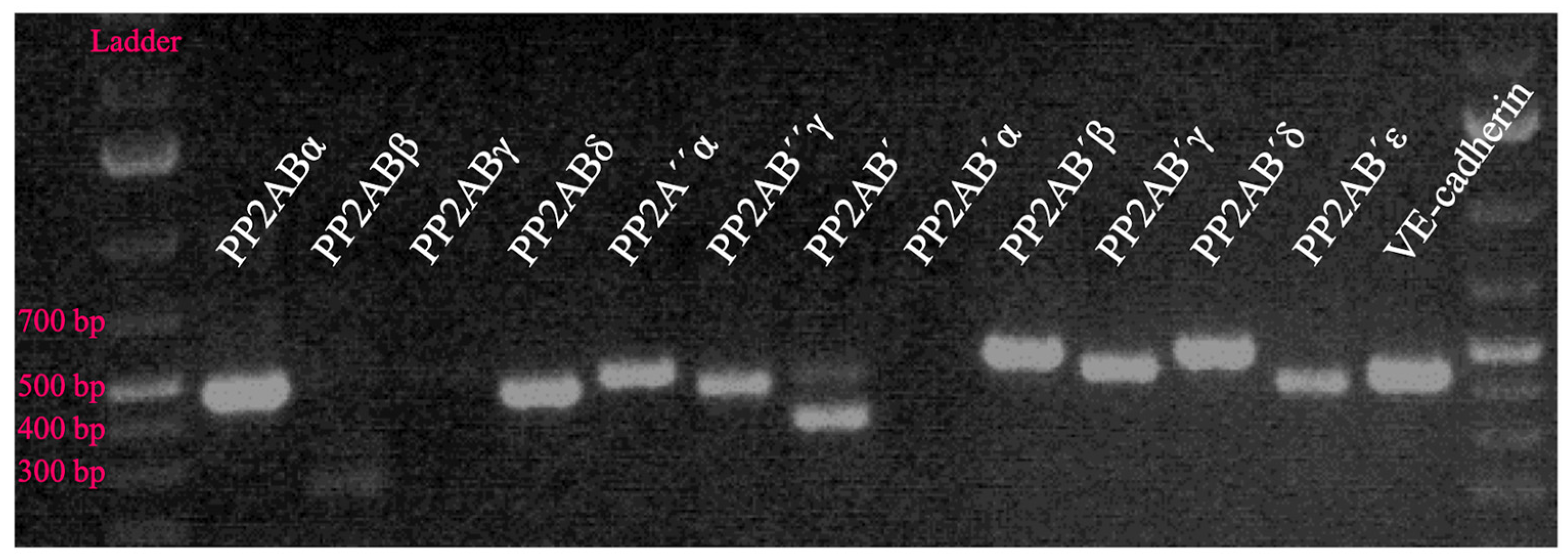
Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
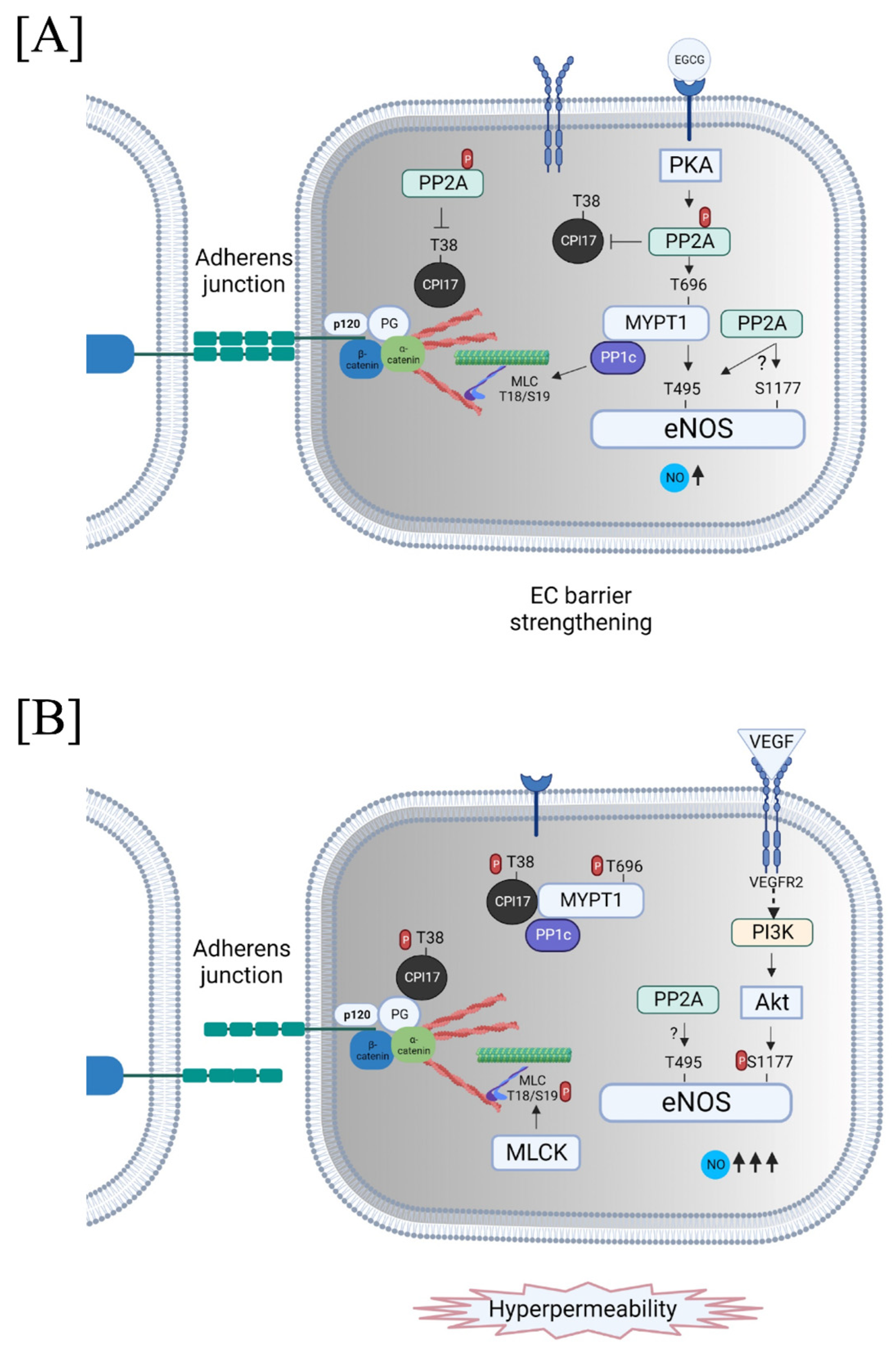{kind=link}
Abstract
:1. Introduction
2. Ser/Thr-Specific Protein Phosphatases, an Overview and Classification
3. Ser/Thr Protein Phosphatase 1
3.1. Structure-Functional Dynamics
3.2. PP1, Cytoskeleton, and EC Barrier Regulation
4. Ser/Thr Protein Phosphatase 2A
4.1. Structure-Functional Dynamics
4.2. PP2A, Cytoskeleton, and EC Barrier Regulation
4.3. Crosstalk between PP2A and MLCP in the Regulation of EC Barrier Function
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Fang, Y.; Wu, D.; Birukov, K.G. Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr. Physiol. 2019, 9, 873–904. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 251–281. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G.N. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kása, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [CrossRef] [Green Version]
- Kugelmann, D.; Rotkopf, L.T.; Radeva, M.Y.; Garcia-Ponce, A.; Walter, E.; Waschke, J. Histamine causes endothelial barrier disruption via Ca2+-mediated RhoA activation and tension at adherens junctions. Sci. Rep. 2018, 8, 13229. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Birukova, A.A. Microtubules as Major Regulators of Endothelial Function: Implication for Lung Injury. Front. Physiol. 2021, 12, 758313. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Verin, A.D. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc. Res. 2008, 76, 202–207. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, E.; Sneh, T.; Moeendarbary, E.; Javanmardi, Y.; Efimova, N.; Yang, C.; Marino-Bravante, G.E.; Chen, X.; Escribano, J.; Spill, F.; et al. Feedback between mechanosensitive signaling and active forces governs endothelial junction integrity. Nat. Commun. 2022, 13, 7089. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Selickman, J.; Vrettou, C.S.; Mentzelopoulos, S.D.; Marini, J.J. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. J. Clin. Med. 2022, 11, 4896. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- McCole, D.F. Phosphatase regulation of intercellular junctions. Tissue Barriers 2013, 1, e26713. [Google Scholar] [CrossRef] [Green Version]
- Csortos, C.; Kolosova, I.; Verin, A.D. Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L843–L854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabutis, N.; Verin, A.; Catravas, J.D. Regulation of pulmonary endothelial barrier function by kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L832–L845. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The regulation of protein function by multisite phosphorylationda 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Johnson, S.A.; Hunter, T. Kinomics: Methods for deciphering the kinome. Nat. Methods 2005, 2, 17–25. [Google Scholar] [CrossRef]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef]
- Zhao, M.-J.; Jiang, H.-R.; Sun, J.-W.; Wang, Z.-A.; Hu, B.; Zhu, C.-R.; Yin, X.-H.; Chen, M.-M.; Ma, X.-C.; Zhao, W.-D.; et al. Roles of RAGE/ROCK1 Pathway in HMGB1-Induced Early Changes in Barrier Permeability of Human Pulmonary Microvascular Endothelial Cell. Front. Immunol. 2021, 12, 697071. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yang, Z.; Yang, C.; Qin, W.; Gu, J.; Hu, C.; Chen, A.; Ning, J.; Yi, B.; Lu, K. A self-organized actomyosin drives multiple intercellular junction disruption and directly promotes neutrophil recruitment in lipopolysaccharide-induced acute lung injury. FASEB J. 2018, 32, 6197–6211. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, N.V.; Zemskova, M.A.; Poirier, C.; Mirzapoiazova, T.; Kolosova, I.; Bresnick, A.R.; Verin, A.D. The suppression of myosin light chain (MLC) phosphorylation during the response to lipopolysaccharide (LPS): Beneficial or detrimental to endothelial barrier? J. Cell. Physiol. 2011, 226, 3132–3146. [Google Scholar] [CrossRef] [Green Version]
- Bogatcheva, N.V.; Dudek, S.M.; Garcia, J.G.; Verin, A.D. Mitogen-activated protein kinases in endothelial pathophysiology. J. Investig. Med. 2003, 51, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Siflinger-Birnboim, A.; Johnson, A. Protein kinase C modulates pulmonary endothelial permeability: A paradigm for acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L435–L451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bátori, R.; Kumar, S.; Bordán, Z.; Cherian-Shaw, M.; Kovács-Kása, A.; MacDonald, J.A.; Fulton, D.J.R.; Erdődi, F.; Verin, A.D. Differential mechanisms of adenosine- and ATPγS-induced microvascular endothelial barrier strengthening. J. Cell. Physiol. 2019, 234, 5863–5879. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Zemskova, M.A.; Kovalenkov, Y.; Poirier, C.; Verin, A.D. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J. Cell. Physiol. 2009, 221, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Wang, Q.; Coughlan, K.; Viollet, B.; Moriasi, C.; Zou, M.-H. Inhibition of AMP-Activated Protein Kinase Accentuates Lipopolysaccharide-Induced Lung Endothelial Barrier Dysfunction and Lung Injury in Vivo. Am. J. Pathol. 2013, 182, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Wojciak-Stothard, B.; Tsang, L.Y.F.; Paleolog, E.; Hall, S.M.; Haworth, S.G. Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1173–L1182. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Hoermann, B.; Kokot, T.; Helm, D.; Heinzlmeir, S.; Chojnacki, J.E.; Schubert, T.; Ludwig, C.; Berteotti, A.; Kurzawa, N.; Kuster, B.; et al. Dissecting the sequence determinants for dephosphorylation by the catalytic subunits of phosphatases PP1 and PP2A. Nat. Commun. 2020, 11, 3583. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, B.; Köhn, M. Evolutionary crossroads of cell signaling: PP1 and PP2A substrate sites in intrinsically disordered regions. Biochem. Soc. Trans. 2021, 49, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Brautigan, D.L.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs-Kasa, A.; Gorshkov, B.A.; Kim, K.-M.; Kumar, S.; Black, S.M.; Fulton, D.J.; Dimitropoulou, C.; Catravas, J.D.; Verin, A.D. The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Sci. Rep. 2016, 6, 39018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-M.; Csortos, C.; Czikora, I.; Fulton, D.; Umapathy, N.S.; Olah, G.; Verin, A.D. Molecular characterization of myosin phosphatase in endothelium. J. Cell. Physiol. 2012, 227, 1701–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kása, A.; Czikora, I.; Verin, A.D.; Gergely, P.; Csortos, C. Protein phosphatase 2A activity is required for functional adherent junctions in endothelial cells. Microvasc. Res. 2013, 89, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Tar, K.; Birukova, A.A.; Csortos, C.; Bakó, E.; Garcia, J.G.; Verin, A.D. Phosphatase 2A is involved in endothelial cell microtubule remodeling and barrier regulation. J. Cell. Biochem. 2004, 92, 534–546. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Ingebritsen, T.S.; Cohen, P. The Protein Phosphatases Involved in Cellular Regulation. 1. Classification and Substrate Specificities. Eur. J. Biochem. 1983, 132, 255–261. [Google Scholar] [CrossRef]
- Virshup, D.M.; Shenolikar, S. From Promiscuity to Precision: Protein Phosphatases Get a Makeover. Mol. Cell 2009, 33, 537–545. [Google Scholar] [CrossRef]
- Heroes, E.; Lesage, B.; Görnemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595. [Google Scholar] [CrossRef]
- Plattner, F.; Bibb, J.A. Serine and Threonine Phosphorylation. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Wayne Albers, R., Price, D.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 467–492. [Google Scholar]
- Casamayor, A.; Ariño, J. Controlling Ser/Thr protein phosphatase PP1 activity and function through interaction with regulatory subunits. Adv. Protein Chem. Struct. Biol. 2020, 122, 231–288. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T.W. Two isoforms of protein phosphatase 1 may be produced from the same gene. FEBS Lett. 1988, 232, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrádi, V.; Axton, J.M.; Brewis, N.D.; da Cruz e Silva, E.F.; Alphey, L.; Cohen, P.T.W. Drosophila contains three genes that encode distinct isoforms of protein phosphatase 1. Eur. J. Biochem. 1990, 194, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Shima, H.; Kitagawa, Y.; Irino, S.; Sugimura, T.; Nagao, M. Identification of members of protein phosphatase 1 gene family int he rat and enhanced expression of protein phosphatase 1 alpha gene in rat hepatocellular carcinomas. Jpn. J. Cancer Res. 1990, 81, 1272–1280. [Google Scholar] [CrossRef]
- Goldberg, J.; Huang, H.-B.; Kwon, Y.-G.; Greengard, P.; Nairn, A.C.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef]
- Kita, A.; Matsunaga, S.; Takai, A.; Kataiwa, H.; Wakimoto, T.; Fusetani, N.; Isobe, M.; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Terrak, M.; Kerff, F.; Langsetmo, K.; Tao, T.; Dominguez, R. Structural basis of protein phosphatase 1 regulation. Nature 2004, 429, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Deng, T.; Xiang, S. Structural basis for protein phosphatase 1 recruitment by glycogen-targeting subunits. FEBS J. 2018, 285, 4646–4659. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Assembly and structure of protein phosphatase 2A. Sci. China Ser. C Life Sci. 2009, 52, 135–146. [Google Scholar] [CrossRef]
- Costas, C.; Louzao, M.C.; Raposo-García, S.; Vale, C.; Vieytes, M.R.; Botana, L.M. Intestinal secretory mechanisms in Okadaic acid induced diarrhoea. Food Chem. Toxicol. 2022, 169, 113449. [Google Scholar] [CrossRef]
- Wang, X.; Obeidat, M.; Li, L.; Pasarj, P.; Aburahess, S.; Holmes, C.F.B.; Ballermann, B.J. TIMAP inhibits endothelial myosin light chain phosphatase by competing with MYPT1 for the catalytic protein phosphatase 1 subunit PP1cβ. J. Biol. Chem. 2019, 294, 13280–13291. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Krizek, J.; Bretscher, A. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics 1992, 132, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guha, S.; Volkert, F.C. The Saccharomyces SHP1 Gene, Which Encodes a Regulator of Phosphoprotein Phosphatase 1 with Differential Effects on Glycogen Metabolism, Meiotic Differentiation, and Mitotic Cell Cycle Progression. Mol. Cell. Biol. 1995, 15, 2037–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peti, W.; Nairn, A.C.; Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013, 280, 596–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, T.D.; Yang, J.; Zhang, L.; Goodwin, K.D.; Zou, Q.; Cortese, M.; Dunker, A.K.; DePaoli-Roach, A.A. Structural Basis for Regulation of Protein Phosphatase 1 by Inhibitor-2. J. Biol. Chem. 2007, 282, 28874–28883. [Google Scholar] [CrossRef] [Green Version]
- Bollen, M. Combinatorial control of protein phosphatase-1. Trends Biochem. Sci. 2001, 26, 426–431. [Google Scholar] [CrossRef]
- Cohen, P.T.W. Protein phosphatase 1–targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [CrossRef]
- Kakinoki, Y.; Kitamura, K.; Matsuzawa, S.-I.; Mizuno, Y.; Miyazaki, T.; Kikuchi, K. Gene expressions and activities of protein phosphatases PP1 and PP2A in rat liver regeneration after partial hepatectomy. Biochem. Biophys. Res. Commun. 1992, 185, 291–297. [Google Scholar] [CrossRef]
- Boudrez, A.; Jagiello, I.; Stalmans, W.; Beullens, M.; Groenen, P.; Van Eynde, A.; Vulsteke, V.; Murray, M.; Krainer, A.R.; Bollen, M. NIPP1-mediated Interaction of Protein Phosphatase-1 with CDC5L, a Regulator of Pre-mRNA Splicing and Mitotic Entry. J. Biol. Chem. 2000, 275, 25411–25417. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, A.; Beullens, M.; Ceulemans, H.; Abt, T.D.; Van Eynde, A.; Nicolaescu, E.; Lesage, B.; Bollen, M. Docking Motif-Guided Mapping of the Interactome of Protein Phosphatase-1. Chem. Biol. 2009, 16, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotto-Lavino, E.; Garcia-Diaz, M.; Du, G.; Frohman, M.A. Basis for the isoformspecific interaction of myosin phosphatase subunits protein phosphatase 1c b and myosin phosphatase targeting subunit 1. J. Biol. Chem. 2010, 285, 6419–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Bertran, M.T.; Mouilleron, S.; Zhou, Y.; Bajaj, R.; Uliana, F.; Kumar, G.S.; van Drogen, A.; Lee, R.; Banerjee, J.J.; Hauri, S.; et al. ASPP proteins discriminate between PP1 catalytic subunits through their SH3 domain and the PP1 C-tail. Nat. Commun. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, S.L.; Domingues, S.C.; da Cruz e Silva, O.A.; Fardilha, M.; da Cruz e Silva, E.F. Protein Phosphatase 1α Interacting Proteins in the Human Brain. Omics 2012, 16, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586, 2732–2739. [Google Scholar] [CrossRef]
- Yadav, L.; Tamene, F.; Göös, H.; van Drogen, A.; Katainen, R.; Aebersold, R.; Gstaiger, M.; Varjosalo, M. Systematic Analysis of Human Protein Phosphatase Interactions and Dynamics. Cell Syst. 2017, 4, 430–444. [Google Scholar] [CrossRef] [Green Version]
- Köhn, M. Turn and Face the Strange: A New View on Phosphatases. ACS Cent. Sci. 2020, 6, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Verin, A.D.; Patterson, C.E.; Day, M.A.; Garcia, J.G. Regulation of endothelial cell gap formation and barrier function by myosin-associated phosphatase activities. Am. J. Physiol. Content 1995, 269, L99–L108. [Google Scholar] [CrossRef]
- Diwan, A.H.; Honkanen, R.E.; Schaeffer, R.C., Jr.; Strada, S.J.; Thompson, W.J. Inhibition of serine-threonine protein phosphatases decreases barrier function of rat pulmonary microvascular endothelial cells. J. Cell. Physiol. 1997, 171, 259–270. [Google Scholar] [CrossRef]
- Favre, B.; Turowski, P.; Hemmings, B.A. Differential Inhibition and Posttranslational Modification of Protein Phosphatase 1 and 2A in MCF7 Cells Treated with Calyculin-A, Okadaic Acid, and Tautomycin. J. Biol. Chem. 1997, 272, 13856–13863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiruppathi, C.; Malik, A.B.; Del Vecchio, P.J.; Keese, C.R.; Giaever, I. Electrical method for detection of endothelial cell shape change in real time: Assessment of endothelial barrier function. Proc. Natl. Acad. Sci. USA 1992, 89, 7919–7923. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Matsuura, N.; Ubukata, M.; Oikawa, H.; Shima, H.; Kikuchi, K. Tautomycetin Is a Novel and Specific Inhibitor of Serine/Threonine Protein Phosphatase Type 1, PP1. Biochem. Biophys. Res. Commun. 2001, 287, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Verin, A.D.; Csortos, C.; Durbin, S.D.; Aydanyan, A.; Wang, P.; Patterson, C.E.; Garcia, J.G. Characterization of the protein phosphatase 1 catalytic subunit in endothelium: Involvement in contractile responses. J. Cell. Biochem. 2000, 79, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Niiro, N.; Hirano, K.; Nishimura, J.; Hartshorne, D.J.; Kanaide, H. Expression, subcellular localization, and cloning of the 130-kDa regulatory subunit of myosin phosphatase in porcine aortic endothelial cells. Biochem. Biophys. Res. Commun. 1999, 254, 490–496. [Google Scholar] [CrossRef]
- Dirksen, W.P.; Vladic, F.; Fisher, S.A. A myosin phosphatase targeting subunit isoform transition defines a smooth muscle developmental phenotypic switch. Am. J. Physiol. Cell Physiol. 2000, 278, C589–C600. [Google Scholar] [CrossRef] [Green Version]
- Xia, D.; Stull, J.T.; Kamm, K.E. Myosin phosphatase targeting subunit 1 affects cell migration by regulating myosin phosphorylation and actin assembly. Exp. Cell Res. 2005, 304, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Ito, M.; Tanaka, J.; Nakano, T.; Kaibuchi, K.; Odai, H.; Takemura, K. Localization of the Gene Coding for Myosin Phosphatase, Target Subunit 1 (MYPT1) to Human Chromosome 12q15–q21. Genomics 1997, 44, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Kirkbride, J.A.; Brautigan, D.L. Assembly of MYPT1 with protein phosphatase-1 in fibroblasts redirects localization and reorganizes the actin cytoskeleton. Cell Motil. Cytoskelet. 2005, 62, 100–109. [Google Scholar] [CrossRef]
- Matsumura, F.; Hartshorne, D.J. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008, 369, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Hartshorne, D.J.; Ito, M.; Erdödi, F. Role of Protein Phosphatase Type 1 in Contractile Functions: Myosin Phosphatase. J. Biol. Chem. 2004, 279, 37211–37214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verin, A.D.; Wang, P.; Garcia, J.G. Immunochemical characterization of myosin-specific phosphatase 1 regulatory subunits in bovine endothelium. J. Cell. Biochem. 2000, 76, 489–498. [Google Scholar] [CrossRef]
- Khromov, A.; Choudhury, N.; Stevenson, A.S.; Somlyo, A.V.; Eto, M. Phosphorylation-dependent Autoinhibition of Myosin Light Chain Phosphatase Accounts for Ca2+ Sensitization Force of Smooth Muscle Contraction. J. Biol. Chem. 2009, 284, 21569–21579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verin, A.D.; Birukova, A.; Wang, P.; Liu, F.; Becker, P.; Birukov, K.; Garcia, J.G.N. Microtubule disassembly increases endothelial cell barrier dysfunction: Role of MLC phosphorylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L565–L574. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Smurova, K.; Birukov, K.G.; Kaibuchi, K.; Garcia, J.G.; Verin, A.D. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc. Res. 2004, 67, 64–77. [Google Scholar] [CrossRef]
- Birukova, A.A.; Smurova, K.; Birukov, K.G.; Usatyuk, P.; Liu, F.; Kaibuchi, K.; Ricks-Cord, A.; Natarajan, V.; Alieva, I.; Garcia, J.G.; et al. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: Role of Rho-dependent mechanisms. J. Cell. Physiol. 2004, 201, 55–70. [Google Scholar] [CrossRef]
- Birukova, A.A.; Birukov, K.G.; Adyshev, D.; Usatyuk, P.; Natarajan, V.; Garcia, J.G.; Verin, A.D. Involvement of microtubules and Rho pathway in TGF-beta1-induced lung vascular barrier dysfunction. J. Cell. Physiol. 2005, 204, 934–947. [Google Scholar] [CrossRef]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin phosphatase: Unexpected functions of a long-known enzyme. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef]
- Butler, T.; Paul, J.; Europe-Finner, N.; Smith, R.; Chan, E.-C. Role of serine-threonine phosphoprotein phosphatases in smooth muscle contractility. Am. J. Physiol. Cell Physiol. 2013, 304, C485–C504. [Google Scholar] [CrossRef] [Green Version]
- Grassie, M.E.; Moffat, L.D.; Walsh, M.P.; MacDonald, J.A. The myosin phosphatase targeting protein (MYPT) family: A regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1δ. Arch. Biochem. Biophys. 2011, 510, 147–159. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Yan, H. Integrin-Linked Kinase Inhibition Attenuates Permeability of the Streptozotocin-Induced Diabetic Rat Retina. Cell Biochem. Biophys. 2013, 67, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, C.; Zhang, H.; Zeng, W.; Li, S.; Chen, C.; Song, X.; Sun, J.; Sun, Z.; Cui, C.; et al. ZIPK mediates endothelial cell contraction through myosin light chain phosphorylation and is required for ischemic-reperfusion injury. FASEB J. 2019, 33, 9062–9074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiller, C.; Mebarek, S.; Jaafar, R.; Pirola, L.; Lagarde, M.; Prigent, A.-F.; Némoz, G. Phospholipase D2 regulates endothelial permeability through cytoskeleton reorganization and occludin downregulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 1236–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A.J. Smooth Muscle Phosphatase Is Regulated in Vivo by Exclusion of Phosphorylation of Threonine 696 of MYPT1 by Phosphorylation of Serine 695 in Response to Cyclic Nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceulemans, H.; Stalmans, W.; Bollen, M. Regulator-driven functional diversification of protein phosphatase-1 in eukaryotic evolution. Bioessays 2002, 24, 371–381. [Google Scholar] [CrossRef]
- Eto, M.; Ohmori, T.; Suzuki, M.; Furuya, K.; Morita, F. A Novel Protein Phosphatase-1 Inhibitory Protein Potentiated by Protein Kinase C. Isolation from Porcine Aorta Media and Characterization1. J. Biochem. 1995, 118, 1104–1107. [Google Scholar] [CrossRef]
- Senba, S.; Eto, M.; Yazawa, M. Identification of Trimeric Myosin Phosphatase (PP1M) as a Target for a Novel PKC-Potentiated Protein Phosphatase-1 Inhibitory Protein (CPI17) in Porcine Aorta Smooth Muscle. J. Biochem. 1999, 125, 354–362. [Google Scholar] [CrossRef]
- Eto, M.; Kitazawa, T.; Brautigan, D.L. Phosphoprotein inhibitor CPI-17 specificity depends on allosteric regulation of protein phosphatase-1 by regulatory subunits. Proc. Natl. Acad. Sci. USA 2004, 101, 8888–8893. [Google Scholar] [CrossRef] [Green Version]
- Eto, M.; Kitazawa, T.; Matsuzawa, F.; Aikawa, S.-I.; Kirkbride, J.A.; Isozumi, N.; Nishimura, Y.; Brautigan, D.L.; Ohki, S.-Y. Phosphorylation-Induced Conformational Switching of CPI-17 Produces a Potent Myosin Phosphatase Inhibitor. Structure 2007, 15, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Eto, M.; Kitazawa, T.; Yazawa, M.; Mukai, H.; Ono, Y.; Brautigan, D.L. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. J. Biol. Chem. 2001, 276, 29072–29078. [Google Scholar] [CrossRef] [Green Version]
- Eto, M. Regulation of Cellular Protein Phosphatase-1 (PP1) by Phosphorylation of the CPI-17 Family, C-kinase-activated PP1 Inhibitors. J. Biol. Chem. 2009, 284, 35273–35277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, M.; Kitazawa, T. Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: Paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. J. Smooth Muscle Res. 2017, 53, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolosova, I.A.; Ma, S.-F.; Adyshev, D.M.; Wang, P.; Ohba, M.; Natarajan, V.; Garcia, J.G.N.; Verin, A.D. Role of CPI-17 in the regulation of endothelial cytoskeleton. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L970–L980. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Chiang, E.T.; Wu, X.; Kelly, G.T.; Kanteti, P.; Singleton, P.A.; Camp, S.M.; Zhou, T.; Dudek, S.M.; Natarajan, V.; et al. Regulation of Thrombin-Induced Lung Endothelial Cell Barrier Disruption by Protein Kinase C Delta. PLoS ONE 2016, 11, e0158865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, P.J.; Bauer, E.M.; Cao, R.; Maniar, S.; Mosher, M.; Chen, J.; Wang, Q.J.; Glorioso, J.C.; Pitt, B.R.; Watkins, S.C.; et al. A role for zinc in regulating hypoxia-induced contractile events in pulmonary endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L874–L886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, M.; Härtel, F.V.; Arshad, M.; Gündüz, D.; Abdallah, Y.; Sauer, H.; Piper, H.M.; Noll, T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: Role of CPI-17. Cardiovasc. Res. 2010, 87, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-M.; Adyshev, D.M.; Kása, A.; Zemskov, E.A.; Kolosova, I.A.; Csortos, C.; Verin, A.D. Putative protein partners for the human CPI-17 protein revealed by bacterial two-hybrid screening. Microvasc. Res. 2013, 88, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, A.S.; Matthew, J.D.; Eto, M.; Luo, S.; Somlyo, A.P.; Somlyo, A.V. Uncoupling of GPCR and RhoA-induced Ca2+-sensitization of chicken amnion smooth muscle lacking CPI-17. FEBS Lett. 2004, 578, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Eto, M.; Kirkbride, J.A.; Chugh, R.; Karikari, N.K.; Kim, J.I. Nuclear localization of CPI-17, a protein phosphatase-1 inhibitor protein, affects histone H3 phosphorylation and corresponds to proliferation of cancer and smooth muscle cells. Biochem. Biophys. Res. Commun. 2013, 434, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Tountas, N.A.; Brautigan, D.L. Migration and retraction of endothelial and epithelial cells require PHI-1, a specific protein-phosphatase-1 inhibitor protein. J. Cell Sci. 2004, 117, 5905–5912. [Google Scholar] [CrossRef] [Green Version]
- Fukata, Y.; Kimura, K.; Oshiro, N.; Saya, H.; Matsuura, Y.; Kaibuchi, K. Association of the Myosin-binding Subunit of Myosin Phosphatase and Moesin: Dual Regulation of Moesin Phosphorylation by Rho-associated Kinase and Myosin Phosphatase. J. Cell Biol. 1998, 141, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Fukata, Y.; Matsuoka, Y.; Bennett, V.; Matsuura, Y.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Regulation of the Association of Adducin with Actin Filaments by Rho-associated Kinase (Rho-kinase) and Myosin Phosphatase. J. Biol. Chem. 1998, 273, 5542–5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogatcheva, N.V.; Zemskova, M.A.; Gorshkov, B.A.; Kim, K.M.; Daglis, G.A.; Poirier, C.; Verin, A.D. Ezrin, Radixin, and Moesin Are Phosphorylated in Response to 2-Methoxyestradiol and Modulate Endothelial Hyperpermeability. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell. Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin is an in vivo substrate for protein kinase C: Phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J. Cell Biol. 1998, 142, 485–497. [Google Scholar] [CrossRef]
- Bretscher, A.; Edwards, K.; Fehon, R.G. ERM proteins and merlin: Integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 2002, 3, 586–599. [Google Scholar] [CrossRef]
- Ivetic, A.; Ridley, A.J. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 2004, 112, 165–176. [Google Scholar] [CrossRef]
- Senju, Y.; Tsai, F.-C. A biophysical perspective of the regulatory mechanisms of ezrin/radixin/moesin proteins. Biophys. Rev. 2022, 14, 199–208. [Google Scholar] [CrossRef]
- Adyshev, D.M.; Dudek, S.M.; Moldobaeva, N.; Kim, K.-M.; Ma, S.-F.; Kasa, A.; Garcia, J.G.N.; Verin, A.D. Ezrin/radixin/moesin proteins differentially regulate endothelial hyperpermeability after thrombin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L240–L255. [Google Scholar] [CrossRef] [Green Version]
- Simó-Servat, O.; Ramos, H.; Bogdanov, P.; García-Ramírez, M.; Huerta, J.; Hernández, C.; Simó, R. ERM Complex, A Therapeutic Target for Vascular Leakage Induced by Diabetes. Curr. Med. Chem. 2022, 29, 2189–2199. [Google Scholar] [CrossRef]
- Koss, M.; Pfeiffer, G.R., 2nd; Wang, Y.; Thomas, S.T.; Yerukhimovich, M.; Gaarde, W.A.; Doerschuk, C.M.; Wang, Q. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J. Immunol. 2006, 176, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-J.; Li, P.-X.; Guo, X.-H.; Huang, Q.-B. Role of moesin, Src, and ROS in advanced glycation end product-induced vascular endothelial dysfunction. Microcirculation 2017, 24, e12358. [Google Scholar] [CrossRef] [PubMed]
- Kiang, K.M.-Y.; Leung, G.K.-K. A Review on Adducin from Functional to Pathological Mechanisms: Future Direction in Cancer. BioMed Res. Int. 2018, 2018, 3465929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin: Structure, function and regulation. Cell. Mol. Life Sci. 2000, 57, 884–895. [Google Scholar] [CrossRef]
- DeOre, B.J.; Partyka, P.P.; Fan, F.; Galie, P.A. CD44 mediates shear stress mechanotransduction in an in vitro blood-brain barrier model through small GTPases RhoA and Rac1. FASEB J. 2022, 36, e22278. [Google Scholar] [CrossRef]
- Lin, Y.; Wozniak, J.M.; Grimsey, N.J.; Girada, S.; Patwardhan, A.; Molinar-Inglis, O.; Smith, T.H.; Lapek, J.D.; Gonzalez, D.J.; Trejo, J. Phosphoproteomic analysis of protease-activated receptor-1 biased signaling reveals unique modulators of endothelial barrier function. Proc. Natl. Acad. Sci. USA 2020, 117, 5039–5048. [Google Scholar] [CrossRef]
- Cao, W.; Mattagajasingh, S.N.; Xu, H.; Kim, K.; Fierlbeck, W.; Deng, J.; Lowenstein, C.J.; Ballermann, B.J. TIMAP, a novel CAAX box protein regulated by TGF-beta1 and expressed in endothelial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C327–C337. [Google Scholar] [CrossRef]
- Boratkó, A.; Csortos, C. TIMAP, the versatile protein phosphatase 1 regulator in endothelial cells. IUBMB Life 2017, 69, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Csortos, C.; Czikora, I.; Bogatcheva, N.V.; Adyshev, D.M.; Poirier, C.; Olah, G.; Verin, A.D. TIMAP is a positive regulator of pulmonary endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L440–L450. [Google Scholar] [CrossRef] [Green Version]
- Poirier, C.; Gorshkov, B.A.; Zemskova, M.A.; Bogatcheva, N.V.; Verin, A.D. TIMAP protects endothelial barrier from LPS-induced vascular leakage and is down-regulated by LPS. Respir. Physiol. Neurobiol. 2011, 179, 334–337. [Google Scholar] [CrossRef] [Green Version]
- Boratkó, A.; Gergely, P.; Csortos, C. RACK1 is involved in endothelial barrier regulation via its two novel interacting partners. Cell Commun. Signal. 2013, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czikora, I.; Kim, K.-M.; Kása, A.; Bécsi, B.; Verin, A.D.; Gergely, P.; Erdődi, F.; Csortos, C. Characterization of the effect of TIMAP phosphorylation on its interaction with protein phosphatase 1. Biochimie 2011, 93, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Kozlowski, K.; Wegner, B.; Rashid, T.; Yeung, T.; Holmes, C.; Ballermann, B.J. Phosphorylation of TIMAP by glycogen synthase kinase-3beta activates its associated protein phosphatase 1. J. Biol. Chem. 2007, 282, 25960–25969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boratkó, A.; Csortos, C. PKC mediated phosphorylation of TIMAP regulates PP1c activity and endothelial barrier function. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Király, N.; Thalwieser, Z.; Fonódi, M.; Csortos, C.; Boratkó, A. Dephosphorylation of annexin A2 by protein phosphatase 1 regulates endothelial cell barrier. IUBMB Life 2021, 73, 1257–1268. [Google Scholar] [CrossRef]
- Boratkó, A.; Péter, M.; Thalwieser, Z.; Kovács, E.; Csortos, C. Elongation factor-1A1 is a novel substrate of the protein phosphatase 1-TIMAP complex. Int. J. Biochem. Cell Biol. 2015, 69, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Shopik, M.J.; Li, L.; Luu, H.-A.; Obeidat, M.; Holmes, C.F.; Ballermann, B.J. Multi-directional function of the protein phosphatase 1 regulatory subunit TIMAP. Biochem. Biophys. Res. Commun. 2013, 435, 567–573. [Google Scholar] [CrossRef]
- Király, N.; Csortos, C.; Boratkó, A. Ser69 phosphorylation of TIMAP affects endothelial cell migration. Exp. Lung Res. 2021, 47, 334–343. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [Green Version]
- Groves, M.R.; Hanlon, N.; Turowski, P.; Hemmings, B.A.; Barford, D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 1999, 96, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cho, U.-S.; Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007, 445, 53–57. [Google Scholar] [CrossRef]
- Sents, W.; Ivanova, E.; Lambrecht, C.; Haesen, D.; Janssens, V. The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J. 2013, 280, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Kashani, E.; Vassella, E. Pleiotropy of PP2A Phosphatases in Cancer with a Focus on Glioblastoma IDH Wildtype. Cancers 2022, 14, 5227. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.; Awal, S.; Vigneron, S.; Roque, S.; Mechali, F.; Labbé, J.C.; Lorca, T.; Castro, A. PP2A-B55: Substrates and regulators in the control of cellular functions. Oncogene 2022, 41, 1–14. [Google Scholar] [CrossRef]
- Kurimchak, A.; Graña, X. PP2A holoenzymes negatively and positively regulate cell cycle progression by dephosphorylating pocket proteins and multiple CDK substrates. Gene 2012, 499, 1–7. [Google Scholar] [CrossRef]
- Lubbers, E.R.; Mohler, P.J. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 2016, 101, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef]
- Alessi, D.; Macdougall, L.K.; Sola, M.M.; Ikebe, M.; Cohen, P. The control of protein phosphatase-1 by targetting subunits. The major myosin phosphatase in avian smooth muscle is a novel form of protein phosphatase-1. Eur. J. Biochem. 1992, 210, 1023–1035. [Google Scholar] [CrossRef]
- Chisholm, A.A.; Cohen, P. The myosin-bound form of protein phosphatase 1 (PP-1M) is the enzyme that dephosphorylates native myosin in skeletal and cardiac muscles. Biochim. Biophys. Acta 1988, 971, 163–169. [Google Scholar]
- Hoffman, A.; Taleski, G.; Sontag, E. The protein serine/threonine phosphatases PP2A, PP1 and calcineurin: A triple threat in the regulation of the neuronal cytoskeleton. Mol. Cell. Neurosci. 2017, 84, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Fowle, H.; Zhao, Z.; Graña, X. PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res. 2019, 144, 55–93. [Google Scholar] [CrossRef]
- Sontag, J.M.; Sontag, E. Regulation of cell adhesion by PP2A and SV40 small tumor antigen: An important link to cell transformation. Cell. Mol. Life Sci. 2006, 63, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Ludueña, R.F. Phosphorylation of beta III-tubulin. Biochemistry 1996, 35, 3704–3711. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Lottes, E.N.; Nanda, S.; Golshir, A.; Patel, A.A.; Ascoli, G.A.; Cox, D.N. PP2A phosphatase regulates cell-type specific cytoskeletal organization to drive dendrite diversity. Front. Mol. Neurosci. 2022, 15, 926567. [Google Scholar] [CrossRef]
- Jackson, J.; Meisinger, J.; Patel, S.; Lim, Z.C.; Vellody, K.; Metz, R.; Young, M.R. Protein phosphatase-2A associates with the cytoskeleton to maintain cell spreading and reduced motility of nonmetastatic Lewis lung carcinoma cells: The loss of this regulatory control in metastatic cells. Invasion Metastasis 1997, 17, 199–209. [Google Scholar]
- Price, N.E.; Wadzinski, B.; Mumby, M.C. An anchoring factor targets protein phosphatase 2A to brain microtubules. Mol. Brain Res. 1999, 73, 68–77. [Google Scholar] [CrossRef]
- Evans, D.R.; Hemmings, B.A. Mutation of the C-terminal leucine residue of PP2Ac inhibits PR55/B subunit binding and confers supersensitivity to microtubule destabilization in Saccharomyces cerevisiae. Mol. Genet. Genom. 2000, 264, 425–432. [Google Scholar] [CrossRef]
- Hiraga, A.; Tamura, S. Protein phosphatase 2A is associated in an inactive state with microtubules through 2A1-specific interaction with tubulin. Biochem. J. 2000, 346, 433–439. [Google Scholar] [CrossRef]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Thurston, V.C.; Zinkowski, R.P.; Binder, L.I. Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma 1996, 105, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.; Tapia, L.; Garrido, J.; Maccioni, R.B. Tau-like Proteins Associated with Centrosomes in Cultured Cells. Exp. Cell Res. 1996, 229, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Ingelson, M.; Vanmechelen, E.; Lannfelt, L. Microtubule-associated protein tau in human fibroblasts with the Swedish Alzheimer mutation. Neurosci. Lett. 1996, 220, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F.; Fellous, A.; McManus, L.; Jordan, M.A.; Nunez, J. Contrasting roles of tau and microtubule-associated protein 2 in the vinblastine-induced aggregation of brain tubulin. J. Biol. Chem. 1984, 259, 12890–12898. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.J.; Grundke-Iqbal, I.; McDonald, B.; Iqbal, K. Comparison of the phosphorylation of microtubule-associated protein tau by non-proline dependent protein kinases. Mol. Cell. Biochem. 1994, 131, 181–189. [Google Scholar] [CrossRef]
- Gupta, R.P.; Abou-Donia, M.B. Tau phosphorylation by diisopropyl phosphorofluoridate (DFP)-treated hen brain supernatant inhibits its binding with microtubules: Role of Ca2+/Calmodulin-dependent protein kinase II in tau phosphorylation. Arch. Biochem. Biophys. 1999, 365, 268–278. [Google Scholar] [CrossRef]
- Litersky, J.M.; Johnson, G.V.W.; Jakes, R.; Goedert, M.; Lee, M.; Seubert, P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem. J. 1996, 316, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Z.; Gong, C.-X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer Paired Helical Filaments by Protein Phosphatase-2A and −2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Taleski, G.; Sontag, E. Protein phosphatase 2A and tau: An orchestrated ‘Pas de Deux’. FEBS Lett. 2018, 592, 1079–1095. [Google Scholar] [CrossRef]
- Gong, C.-X.; Wegiel, J.; Lidsky, T.; Zuck, L.; Avila, J.; Wisniewski, H.M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of phosphorylation of neuronal microtubule-associated proteins MAP1b and MAP2 by protein phosphatase-2A and -2B in rat brain. Brain Res. 2000, 853, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Schmidt, G.; Tompa, P.; Ogueta, S.; Vázquez, J.; Kulcsár, P.; Kovács, J.; Dombrádi, V.; Friedrich, P. The Phosphorylation State of Threonine-220, a Uniquely Phosphatase-Sensitive Protein Kinase A Site in Microtubule-Associated Protein MAP2c, Regulates Microtubule Binding and Stability. Biochemistry 2002, 41, 12427–12435. [Google Scholar] [CrossRef] [PubMed]
- Sontag, J.M.; Nunbhakdi-Craig, V.; White, C.L., 3rd; Halpain, S.; Sontag, E. The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: Implications for tauopathies. J. Biol. Chem. 2012, 287, 14984–14993. [Google Scholar] [CrossRef] [Green Version]
- Bogatcheva, N.V.; Adyshev, D.; Mambetsariev, B.; Moldobaeva, N.; Verin, A.D. Involvement of microtubules, p38, and Rho kinases pathway in 2-methoxyestradiol-induced lung vascular barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L487–L499. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Ke, Y.; Tian, Y.; Ohmura, T.; Sitikov, A.; Sarich, N.; Montgomery, C.P.; Birukova, A.A. Staphylococcus aureus–induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 2019, 294, 3369–3384. [Google Scholar] [CrossRef] [Green Version]
- Gorshkov, B.A.; Zemskova, M.A.; Verin, A.D.; Bogatcheva, N.V. Taxol alleviates 2-methoxyestradiol-induced endothelial permeability. Vasc. Pharmacol. 2012, 56, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Mirzapoiazova, T.; Kolosova, I.A.; Moreno, L.; Sammani, S.; Garcia, J.G.N.; Verin, A.D. Suppression of endotoxin-induced inflammation by taxol. Eur. Respir. J. 2007, 30, 429–435. [Google Scholar] [CrossRef]
- Tian, X.; Tian, Y.; Moldobaeva, N.; Sarich, N.; Birukova, A.A. Microtubule Dynamics Control HGF-Induced Lung Endothelial Barrier Enhancement. PLoS ONE 2014, 9, e105912. [Google Scholar] [CrossRef] [Green Version]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large Potentials of Small Heat Shock Proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef] [Green Version]
- Muranova, L.K.; Shatov, V.M.; Gusev, N.B. Role of Small Heat Shock Proteins in the Remodeling of Actin Microfilaments. Biochemistry 2022, 87, 800–811. [Google Scholar] [CrossRef]
- Hino, M.; Kurogi, K.; Okubo, M.A.; Murata-Hori, M.; Hosoya, H. Small heat shock protein 27 (HSP27) associates with tubu-lin/microtubules in HeLa cells. Biochem. Biophys. Res. Commun. 2000, 271, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Sun, Y.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR induces cytoskeleton system reorganization through hyperphos-phorylation of tau and HSP27 via PP2A inhibition and subsequent activation of the p38 MAPK signaling pathway in neuroendocrine (PC12) cells. Toxicology 2011, 290, 218–229. [Google Scholar] [CrossRef]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.; Houle, F.; Rousseau, S.; Deschesnes, R.G.; Shah, G.M.; Landry, J. SAPK2/p38-dependent F-Actin Reorganization Regulates Early Membrane Blebbing during Stress-induced Apoptosis. J. Cell Biol. 1998, 143, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, C.; Ross, S.E.; Bragado, M.J.; Groblewski, G.E.; Ernst, S.A.; Williams, J.A. A Role for the p38 Mitogen-activated Protein Kinase/Hsp 27 Pathway in Cholecystokinin-induced Changes in the Actin Cytoskeleton in Rat Pancreatic Acini. J. Biol. Chem. 1998, 273, 24173–24180. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, C.; Clapp, P.; Welsh, M.J.; Benndorf, R.; Williams, J.A. HSP27 expression regulates CCK-induced changes of the actin cytoskeleton in CHO-CCK-A cells. Am. J. Physiol. Cell Physiol. 1999, 277, C1032–C1043. [Google Scholar] [CrossRef]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Piotrowicz, R.S.; Levin, E.G. Basolateral Membrane-associated 27-kDa Heat Shock Protein and Microfilament Polymerization. J. Biol. Chem. 1997, 272, 25920–25927. [Google Scholar] [CrossRef] [Green Version]
- Rada, C.C.; Mejia-Pena, H.; Grimsey, N.J.; Cordova, I.C.; Olson, J.; Wozniak, J.M.; Gonzalez, D.J.; Nizet, V.; Trejo, J. Heat shock protein 27 activity is linked to endothelial barrier recovery after proinflammatory GPCR-induced disruption. Sci. Signal. 2021, 14, eabc1044. [Google Scholar] [CrossRef]
- Sun, H.-B.; Ren, X.; Liu, J.; Guo, X.-W.; Jiang, X.-P.; Zhang, D.-X.; Huang, Y.-S.; Zhang, J.-P. HSP27 phosphorylation protects against endothelial barrier dysfunction under burn serum challenge. Biochem. Biophys. Res. Commun. 2015, 463, 377–383. [Google Scholar] [CrossRef]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Liu, J.; Tian, C.; Zeng, Y.; Zheng, Y.-H.; Fang, Q.; Li, H.-H. Epigallocatechin gallate inhibits angiotensin II-induced endothelial barrier dysfunction via inhibition of the p38 MAPK/HSP27 pathway. Acta Pharmacol. Sin. 2010, 31, 1401–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, S.; Rees, R.S.; Yancy, S.L.; Welsh, M.J.; Remick, D.G.; Yamada, T.; Hata, J.; Gilmont, R.R. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell Biol. Toxicol. 2004, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.; Qin, S.; Philp, R.; Tan, Y.H.; Guy, G.R. Dephosphorylation of the small heat shock protein Hsp27 in vivo by protein phosphatase 2A. J. Biol. Chem. 1994, 269, 9176–9183. [Google Scholar] [CrossRef] [PubMed]
- Berrou, E.; Bryckaert, M. Recruitment of protein phosphatase 2A to dorsal ruffles by platelet-derived growth factor in smooth muscle cells: Dephosphorylation of Hsp27. Exp. Cell Res. 2009, 315, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.B.; Bitar, K.N. RhoA- and PKC-α-mediated phosphorylation of MYPT and its association with HSP27 in colonic smooth muscle cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G83–G95. [Google Scholar] [CrossRef]
- Armstrong, S.C.; Delacey, M.; Ganote, C.E. Phosphorylation State of hsp27 and p38 MAPK During Preconditioning and Protein Phosphatase Inhibitor Protection of Rabbit Cardiomyocytes. J. Mol. Cell. Cardiol. 1999, 31, 555–567. [Google Scholar] [CrossRef]
- Nunbhakdi-Craig, V.; Craig, L.; Machleidt, T.; Sontag, E. Simian Virus 40 Small Tumor Antigen Induces Deregulation of the Actin Cytoskeleton and Tight Junctions in Kidney Epithelial Cells. J. Virol. 2003, 77, 2807–2818. [Google Scholar] [CrossRef] [Green Version]
- Nita-Lazar, M.; Rebustini, I.; Walker, J.; Kukuruzinska, M.A. Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp. Cell Res. 2010, 316, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Schuhmacher, D.; Sontag, J.-M.; Sontag, E. Protein Phosphatase 2A: More Than a Passenger in the Regulation of Epithelial Cell–Cell Junctions. Front. Cell Dev. Biol. 2019, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Geudens, I.; Bruyr, J.; Potente, M.; Bleuart, A.; Lebrun, M.; Simonis, N.; Deroanne, C.; Twizere, J.-C.; Soubeyran, P.; et al. PP2A regulatory subunit Bα controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J. 2013, 32, 2491–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Bei, Y.; Lin, H.; Wei, T.; Dai, Y.; Hu, Y.; Zhang, C.; Dai, H. The role of class IIa histone deacetylases in regulating endothelial function. Front. Physiol. 2023, 14, 1091794. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorganic Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef]
- Kovacs-Kasa, A.; Kovacs, L.; Cherian-Shaw, M.; Patel, V.; Meadows, M.L.; Fulton, D.J.; Su, Y.; Verin, A.D. Inhibition of Class IIa HDACs improves endothelial barrier function in endotoxin-induced acute lung injury. J. Cell. Physiol. 2021, 236, 2893–2905. [Google Scholar] [CrossRef]
- Young, M.R.I.; Kolesiak, K.; Meisinger, J. Protein phosphatase-2A regulates endothelial cell motility and both the phosphorylation and the stability of focal adhesion complexes. Int. J. Cancer 2002, 100, 276–282. [Google Scholar] [CrossRef]
- Ripamonti, M.; Wehrle-Haller, B.; de Curtis, I. Paxillin: A Hub for Mechano-Transduction from the β3 Integrin-Talin-Kindlin Axis. Front. Cell Dev. Biol. 2022, 10, 852016. [Google Scholar] [CrossRef]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Alekseeva, E.; Cokic, I.; Turner, C.E.; Birukov, K.G.; Tian, Y.; Tian, X.; Gawlak, G.; Sarich, N.; Sacks, D.B.; et al. Cross talk between paxillin and Rac is critical for mediation of barrier-protective effects by oxidized phospholipids. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L593–L602. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Iwasaki, Y.; Yamada, S.; Kamiguchi, H.; Sakakibara, S.-I. Control of cell migration by the novel protein phosphatase-2A interacting protein inka2. Cell Tissue Res. 2020, 380, 527–537. [Google Scholar] [CrossRef]
- Yeh, P.-A.; Chang, C.-J. A novel function of twins, B subunit of protein phosphatase 2A, in regulating actin polymerization. PLoS ONE 2017, 12, e0186037. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.J.; Huxley, V.H. Capillary hydraulic conductivity is elevated by cGMP-dependent vasodilators. Circ. Res. 1992, 70, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.S.; Villablanca, A.C.; Rutledge, J.C. Substance P increases microvascular permeability via nitric oxide-mediated convective pathways. Am. J. Physiol. Content 1995, 268, R1060–R1068. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, M.M.; Quardt, S.M.; Kim, D.; Oshiro, H.; Minnicozzi, M.; Durán, W.N. Platelet Activating Factor Modulates Microvascular Permeability through Nitric Oxide Synthesis. Microvasc. Res. 1995, 50, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Huang, Q.B.; Yuan, Y.; Granger, H.J. VEGF induces NO-dependent hyperpermeability in coronary venules. Am. J. Physiol. Circ. Physiol. 1996, 271, H2735–H2739. [Google Scholar] [CrossRef]
- Durán, W.N.; Breslin, J.W.; Sánchez, F.A. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc. Res. 2010, 87, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Durán, W.N.; Beuve, A.V.; Sánchez, F.A. Nitric oxide, S-nitrosation, and endothelial permeability. IUBMB Life 2013, 65, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Ninchoji, T.; Love, D.T.; Smith, R.O.; Hedlund, M.; Vestweber, D.; Sessa, W.C.; Claesson-Welsh, L. eNOS-induced vascular barrier disruption in retinopathy by c-Src activation and tyrosine phosphorylation of VE-cadherin. eLife 2021, 10, e64944. [Google Scholar] [CrossRef]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharmacol. 2019, 176, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.L.; Sessa, W.C. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 2013, 126, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Heiss, E.H.; Dirsch, V.M. Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 2014, 20, 3503–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, M.; Nakayama, M.; Goto, T.M.; Amano, M.; Komori, K.; Kaibuchi, K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem. Biophys. Res. Commun. 2007, 361, 462–467. [Google Scholar] [CrossRef]
- Michell, B.J.; Chen, Z.P.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greif, D.M.; Kou, R.; Michel, T. Site-Specific Dephosphorylation of Endothelial Nitric Oxide Synthase by Protein Phosphatase 2A: Evidence for Crosstalk between Phosphorylation Sites. Biochemistry 2002, 41, 15845–15853. [Google Scholar] [CrossRef]
- Bátori, R.; Bécsi, B.; Nagy, D.; Kónya, Z.; Hegedűs, C.; Bordán, Z.; Verin, A.; Lontay, B.; Erdődi, F. Interplay of myosin phosphatase and protein phosphatase-2A in the regulation of endothelial nitric-oxide synthase phosphorylation and nitric oxide production. Sci. Rep. 2017, 7, 44698. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, N.; Niiro, N.; Ikebe, M. Dephosphorylation of the two regulatory components of myosin phosphatase, MBS and CPI17. FEBS Lett. 2002, 515, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Matos, B.; Howl, J.; Jerónimo, C.; Fardilha, M. Modulation of serine/threonine-protein phosphatase 1 (PP1) complexes: A promising approach in cancer treatment. Drug Discov. Today 2021, 26, 2680–2698. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, R.S.; Kovacs-Kasa, A.; Gorshkov, B.A.; Fulton, D.J.R.; Su, Y.; Batori, R.K.; Verin, A.D. Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation. Biomedicines 2023, 11, 1638. https://doi.org/10.3390/biomedicines11061638
Patil RS, Kovacs-Kasa A, Gorshkov BA, Fulton DJR, Su Y, Batori RK, Verin AD. Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation. Biomedicines. 2023; 11(6):1638. https://doi.org/10.3390/biomedicines11061638
Chicago/Turabian StylePatil, Rahul S., Anita Kovacs-Kasa, Boris A. Gorshkov, David J. R. Fulton, Yunchao Su, Robert K. Batori, and Alexander D. Verin. 2023. "Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation" Biomedicines 11, no. 6: 1638. https://doi.org/10.3390/biomedicines11061638
APA StylePatil, R. S., Kovacs-Kasa, A., Gorshkov, B. A., Fulton, D. J. R., Su, Y., Batori, R. K., & Verin, A. D. (2023). Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation. Biomedicines, 11(6), 1638. https://doi.org/10.3390/biomedicines11061638