

Wall Tension and Tubular Resistance in Kidney Cystic Conditions


Abstract
:1. Introduction
ADPKD Cystogenesis
- Increased fluid secretion from tubular cells. It has been observed that the cyst fluid from patients with ADPKD has effects on cell secretion in vitro [9]. This led to the hypothesis that fluid secretion was important for cyst formation. Furthermore, two subjects with both cystic fibrosis (which should induce altered tubular secretion) and ADPKD were noted to have a milder kidney phenotype. This observation was difficult to confirm [10]. The hypothesis was that the anomalous transmembrane conductance regulator (CFTR) in cystic fibrosis could modulate cyst expansion. Within the same direction, another hypothesis suggests that epithelial cells in ADPKD have altered cyclic AMP, and this increases secretion and, therefore, cyst formation. Overall, this secretion hypothesis does not explain the extrarenal “signatures” of ADPKD (see below).
- Epithelial cell proliferation. Cyst fluid from patients with ADPKD does not only change cell secretion but also cell proliferation in vitro [9]. It is unclear if this also happens in humans and in vivo. However, the research has focused on possible mediators of cell proliferation, such as mammalian target of rapamycin (mTOR), tuberin [11], a shift to aerobic glycolysis [12,13], JAK-STAT, IL-13, STAT6 [14], EGFR/EGF, and TGFa [15]. ERK is also involved in cyst expansion and cellular proliferation by its association with the scaffolding protein PEA15 [16,17]. The major problem of this theory is that cell proliferation is more likely an effect of cyst enlargement rather that its cause, as discussed below. Furthermore, it does not explain the extrarenal “signatures” of ADPKD (see below).
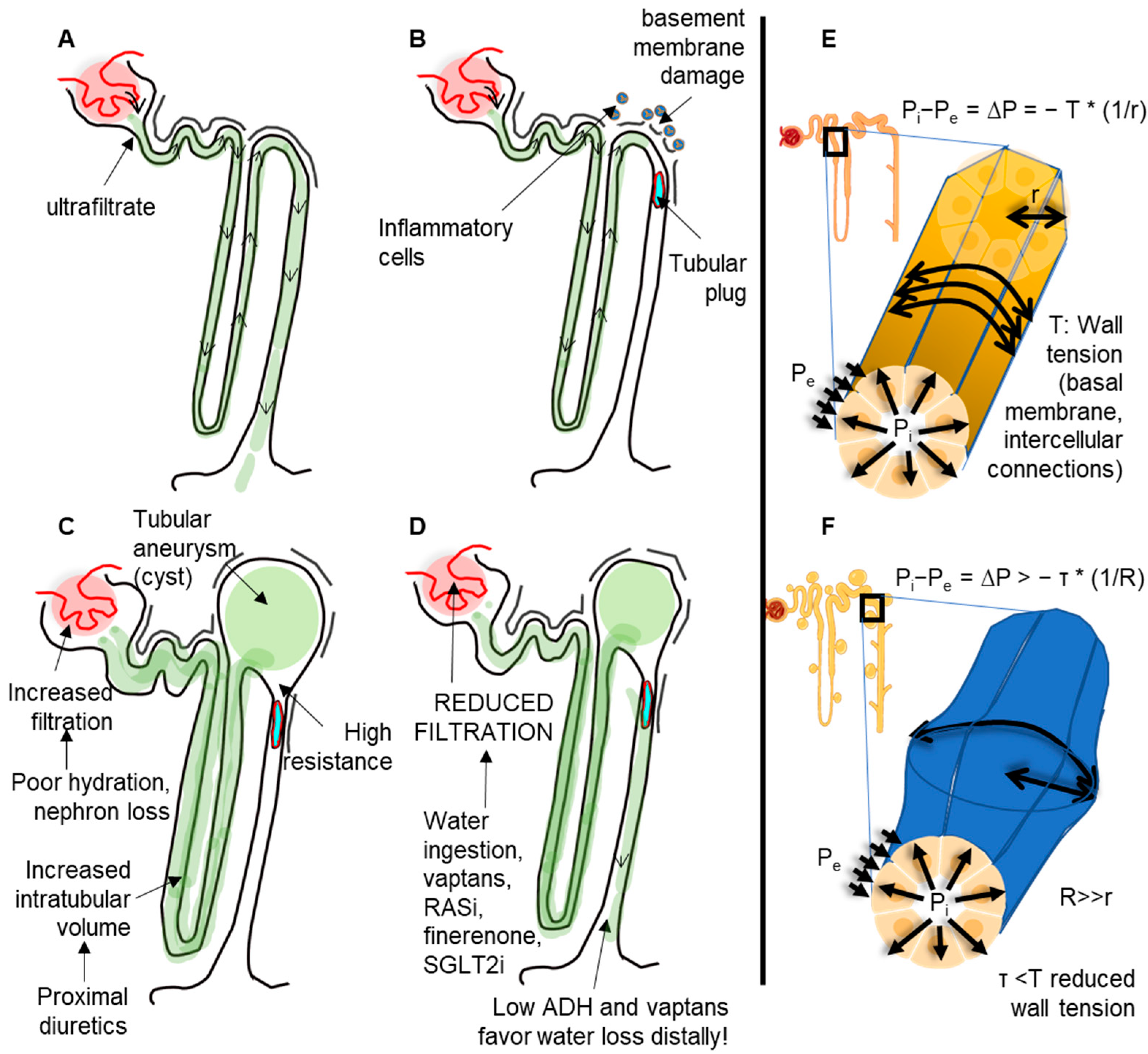
2. Laplace’s Law in Kidney Tubules: Hypothesis on ADPKD
3. ADPKD: Major Extrarenal Features
- Pancreas: Patients with ADPKD can show both cysts in pancreatic tissue and, with lower frequency, intraductal papillary mucinous neoplasms (IPMN) [57].
- Male reproductive organs: Cysts have frequently been observed (43% of ADPKD cases) in the rete testis, epididymis, seminal vesicles, and prostate [58].
- Abdominal wall: Patients with ADPKD may often present with abdominal wall hernias (45%) [61]. Although the mass effect caused by the enlargement of the kidneys may contribute to this phenotype, a reduced wall tension may also be responsible. Indeed, the placements of catheters, such as in peritoneal dialysis, almost doubles the prevalence of hernias in subjects with ADPKD compared to those without ADPKD [62,63]. Furthermore, nephrectomy, which should lower intra-abdominal pressure, is also associated with incisional hernias [64].
- Ovarian cysts: Single ovarian cysts are frequent in ADPKD [65].
- Lungs: Bronchiectasis has been described [66].
- Brain: No gross brain abnormalities have been described in patients with ADPKD. However, they might suffer from depression [67]. Interestingly, no mild cognitive impairment has been reported in these patients, although it is often present in other forms of CKD [67,68,69,70,71,72,73]. Both PKD1 and PKD2 have large expression in the brain (data from Allen Brain Atlas database and from Human Protein Atlas database). Therefore, their absence may be expected to lead to hyperactivity [74], anxiety [75], and memory disturbances [76].
- Cancer: At present, the role of ADPKD in renal cancer is controversial [31].
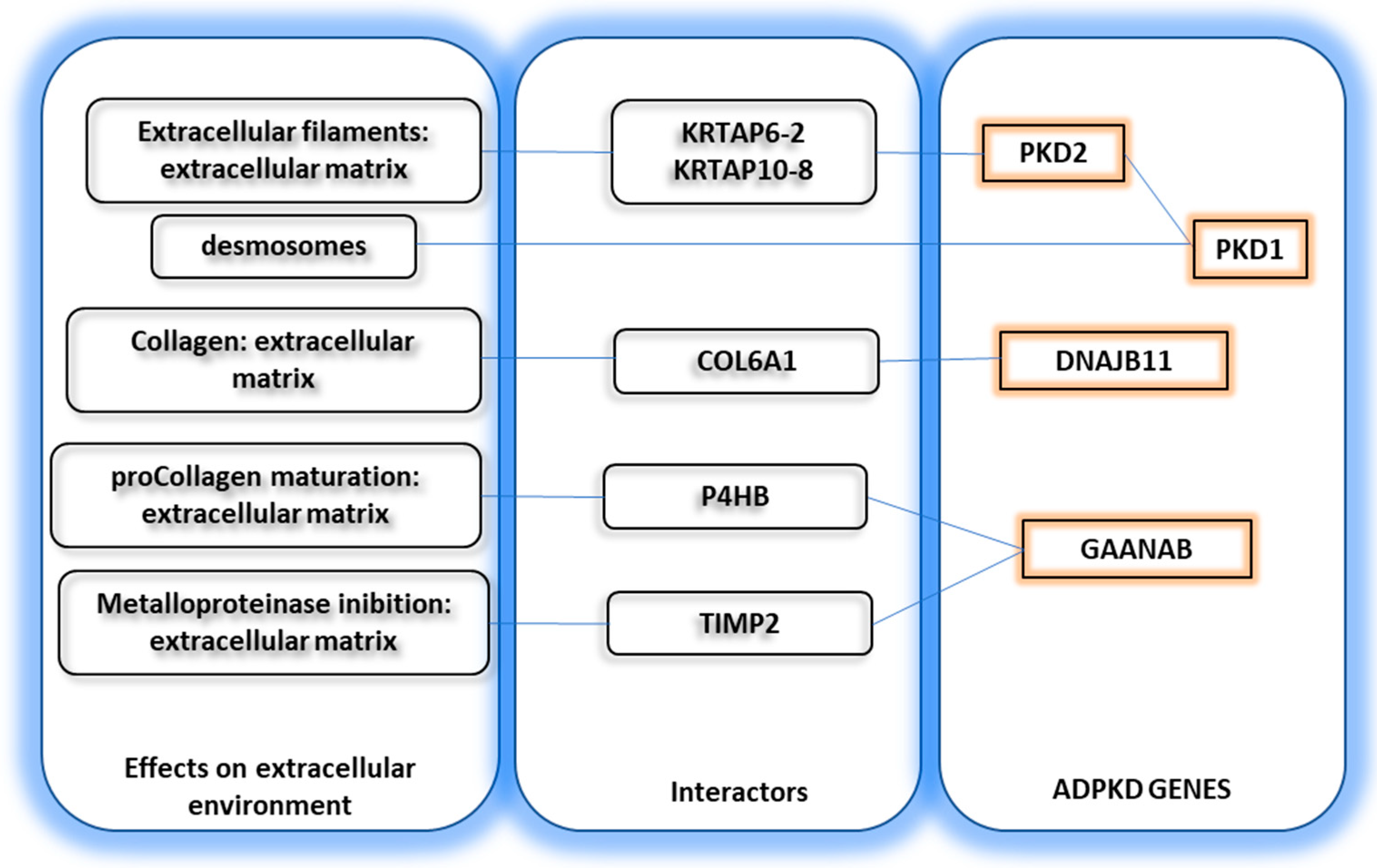
4. Putative Molecular Links between Polycystin Proteins and Tubule Wall Tension
5. Acquired Cystic Kidney Disease (ACKD): A Unified View
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balat, A. Tear drops of kidney: A historical overview of Polycystic Kidney Disease. G. Ital. Nefrol. 2016, 33 (Suppl. S6), 1724–5590. [Google Scholar]
- Bell, E. A Classification of Renal Tumors with Observations on the Frequency of the Various Types. J. Urol. 1938, 39, 238–243. [Google Scholar]
- Perna, A.F.; Zacchia, M.; Trepiccione, F.; Ingrosso, D. The Sulfur Metabolite Lanthionine: Evidence for a Role as a Novel Uremic Toxin. Toxins 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Pizza, A.; Di Nunzio, A.; Bellantone, R.; Raffaelli, M.; Cicchella, T.; Conzo, G.; Santini, L.; Zacchia, M.; Trepiccione, F.; et al. ADAM17, a New Player in the Pathogenesis of Chronic Kidney Disease-Mineral and Bone Disorder. J. Ren. Nutr. 2017, 27, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, G.; Gryp, T.; Perna, A. Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins 2020, 12, 245. [Google Scholar] [CrossRef] [Green Version]
- Viggiano, D.; Gigliotti, G.; Vallone, G.; Giammarino, A.; Nigro, M.; Capasso, G. Urate-Lowering Agents in Asymptomatic Hyperuricemia: Role of Urine Sediment Analysis and Musculoskeletal Ultrasound. Kidney Blood Press. Res. 2018, 43, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Cuppage, F.E.; Huseman, R.A.; Chapman, A.; Grantham, J.J. Ultrastructure and function of cysts from human adult polycystic kidneys. Kidney Int. 1980, 17, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Grantham, J.J.; Donoso, V.S.; Evan, A.P.; Carone, F.A.; Gardner, K.D. Viscoelastic properties of tubule basement membranes in experimental renal cystic disease. Kidney Int. 1987, 32, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Nagao, S.; Takahashi, H.; Ye, M.; Grantham, J.J. Cyst fluid from a murine model of polycystic kidney disease stimulates fluid secretion, cyclic adenosine monophosphate accumulation, and cell proliferation by Madin-Darby canine kidney cells in vitro. Am. J. Kidney Dis. 1995, 25, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Glockner, J.F.; Rossetti, S.; Babovich-Vuksanovic, D.; Harris, P.C.; Torres, V.E. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J. Nephrol. 2006, 19, 529–534. [Google Scholar]
- Harris, P.C. 2008 Homer, W. Smith Award: Insights into the pathogenesis of polycystic kidney disease from gene discovery. J. Am. Soc. Nephrol. 2009, 20, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Borriello, M.; Liccardo, M.; Scafuro, M.; Russo, P.; Iannuzzi, C. Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants 2021, 10, 1127. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Olsan, E.E.; Mukherjee, S.; Wulkersdorfer, B.; Shillingford, J.M.; Giovannone, A.J.; Todorov, G.; Song, X.; Pei, Y.; Weimbs, T. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2011, 108, 18067–18072. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wilson, P.D. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am. J. Physiol. 1995, 269, C487–C495. [Google Scholar] [CrossRef]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Iovino, S.; Oriente, F.; Botta, G.; Cabaro, S.; Iovane, V.; Paciello, O.; Viggiano, D.; Perruolo, G.; Formisano, P.; Beguinot, F. PED/PEA-15 induces autophagy and mediates TGF-beta1 effect on muscle cell differentiation. Cell Death Differ. 2012, 19, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Bracken, C.; Beauverger, P.; Duclos, O.; Russo, R.J.; Rogers, K.A.; Husson, H.; Natoli, T.A.; Ledbetter, S.R.; Janiak, P.; Ibraghimov-Beskrovnaya, O.; et al. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am. J. Physiol. Physiol. 2016, 310, F1414–F1422. [Google Scholar] [CrossRef] [Green Version]
- Zlomuzica, A.; Viggiano, D.; Degen, I.; Binder, S.; Ruocco, L.A.; Sadile, A.G.A.; Willecke, K.; Huston, J.J.P.; Dere, E.; Degen, J.; et al. Behavioral alterations and changes in Ca/calmodulin kinase II levels in the striatum of connexin36 deficient mice. Behav. Brain Res. 2012, 226, 293–300. [Google Scholar] [CrossRef]
- Fischer, E.; Legue, E.; Doyen, A.; Nato, F.; Nicolas, J.-F.; Torres, V.; Yaniv, M.; Pontoglio, M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006, 38, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Saburi, S.; Hester, I.; Fischer, E.; Pontoglio, M.; Eremina, V.; Gessler, M.; Quaggin, S.E.; Harrison, R.; Mount, R.; McNeill, H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 2008, 40, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, V.; Qian, F.; Boute, N.; Cai, Y.; Phakdeekitacharoen, B.; Onuchic, L.F.; Attié-Bitach, T.; Guicharnaud, L.; Devuyst, O.; Germino, G.G.; et al. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am. J. Pathol. 2002, 160, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-Y.; Ong, A.C.M. Mechanism-based therapeutics for autosomal dominant polycystic kidney disease: Recent progress and future prospects. Nephron. Clin. Pract. 2012, 120, c25–c34; discussion c35. [Google Scholar] [CrossRef]
- Perico, N.; Ruggenenti, P.; Perna, A.; Caroli, A.; Trillini, M.; Sironi, S.; Pisani, A.; Riccio, E.; Imbriaco, M.; Dugo, M.; et al. Octreotide-LAR in later-stage autosomal dominant polycystic kidney disease (ALADIN 2): A randomized, double-blind, placebo-controlled, multicenter trial. PLoS Med. 2019, 16, e1002777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Aerts, R.M.M.; Kievit, W.; D’Agnolo, H.M.A.; Blijdorp, C.J.; Casteleijn, N.F.; Dekker, S.E.I.; de Fijter, J.W.; van Gastel, M.; Gevers, T.J.; van de Laarschot, L.F.M.; et al. Lanreotide Reduces Liver Growth In Patients With Autosomal Dominant Polycystic Liver and Kidney Disease. Gastroenterology 2019, 157, 481–491.e7. [Google Scholar] [CrossRef]
- Hatzoglou, A.; Bakogeorgou, E.; Papakonstanti, E.; Stournaras, C.; Emmanouel, D.S.; Castanas, E. Identification and characterization of opioid and somatostatin binding sites in the opossum kidney (OK) cell line and their effect on growth. J. Cell. Biochem. 1996, 63, 410–421. [Google Scholar] [CrossRef]
- Grantham, J.J.; Geiser, J.L.; Evan, A.P. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987, 31, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Gilmer, G.G.; Deshpande, V.G.; Chou, C.-L.; Knepper, M. Flow resistance along the rat renal tubule. Am. J. Physiol. Physiol. 2018, 315, F1398–F1405. [Google Scholar] [CrossRef]
- Capasso, A.; Benigni, A.; Capitanio, U.; Danesh, F.R.F.; Di Marzo, V.; Gesualdo, L.; Grandaliano, G.; Jaimes, E.A.E.; Malyszko, J.; Perazella, M.A.; et al. Summary of the International Conference on Onco-Nephrology: An emerging field in medicine. Kidney Int. 2019, 9, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, F.; Brasseur, J.G. The invalidity of the Laplace law for biological vessels and of estimating elastic modulus from total stress vs. strain: A new practical method. Math. Med. Biol. 2015, 32, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Bae, K.T.; Zhou, W.; Shen, C.; Landsittel, D.P.; Wu, Z.; Tao, C.; Chapman, A.B.; Torres, V.E.; Yu, A.S.L.; Mrug, M.; et al. Growth Pattern of Kidney Cyst Number and Volume in Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Masella, C.; Viggiano, D.; Molfino, I.; Zacchia, M.; Capolongo, G.; Anastasio, P.; Simeoni, M. Diuretic Resistance in Cardio-Nephrology: Role of Pharmacokinetics, Hypochloremia, and Kidney Remodeling. Kidney Blood Press. Res. 2019, 44, 915–927. [Google Scholar] [CrossRef]
- Capolongo, G.; Capasso, G.; Viggiano, D. A Shared Nephroprotective Mechanism for Co-Transporter 2 Inhibitors, and Vasopressin Receptor Antagonists: Immunology Meets Hemodynamics. Int. J. Mol. Sci. 2022, 23, 3915. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, H.; Yang, B. Drug discovery for polycystic kidney disease. Acta Pharmacol. Sin. 2011, 32, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Jiang, S.-T.; Chiou, Y.-Y.; Wang, E.; Lin, H.-K.; Lin, Y.-T.; Chi, Y.-C.; Wang, C.-K.L.; Tang, M.-J.; Li, H. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am. J. Pathol. 2006, 168, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y.; Sawa, N.; Hasegawa, E.; Mizuno, H.; Kawada, M.; Sekine, A.; Hiramatsu, R.; Yamanouchi, M.; Hayami, N.; Suwabe, T.; et al. PKD1-associated autosomal dominant polycystic kidney disease with glomerular cysts presenting with nephrotic syndrome caused by focal segmental glomerulosclerosis. BMC Nephrol. 2019, 20, 337. [Google Scholar] [CrossRef]
- Verani, R.R.; Silva, F.G. Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: A histochemical study. Mod. Pathol. 1988, 1, 457–463. [Google Scholar] [PubMed]
- Wu, G.; D’Agati, V.; Cai, Y.; Markowitz, G.; Park, J.H.; Reynolds, D.M.; Maeda, Y.; Le, T.C.; Hou, H.; Kucherlapati, R.; et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 1998, 93, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, K.D.; Burnside, J.S.; Skipper, B.J.; Swan, S.K.; Bennett, W.M.; Connors, B.A.; Evan, A.P. On the probability that kidneys are different in autosomal dominant polycystic disease. Kidney Int. 1992, 42, 1199–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galarreta, C.I.; Grantham, J.J.; Forbes, M.S.; Maser, R.L.; Wallace, D.P.; Chevalier, R.L. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am. J. Pathol. 2014, 184, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, W.N.; Happe, H.; Peters, D.J.M. Variable Cyst Development in Autosomal Dominant Polycystic Kidney Disease: The Biologic Context. J. Am. Soc. Nephrol. 2016, 27, 3530–3538. [Google Scholar] [CrossRef] [Green Version]
- Grantham, J.J.; Mulamalla, S.; Grantham, C.J.; Wallace, D.P.; Cook, L.T.; Wetzel, L.H.; Fields, T.A.; Bae, K.T. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin. J. Am. Soc. Nephrol. 2012, 7, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume Progression in Polycystic Kidney Disease. N. Engl. J. Med. 2006, 354, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Milutinovic, J.; Fialkow, P.J.; Rudd, T.G.; Agodoa, L.Y.; Phillips, L.A.; Bryant, J.I. Liver cysts in patients with autosomal dominant polycystic kidney disease. Am. J. Med. 1980, 68, 741–744. [Google Scholar] [CrossRef]
- Masyuk, T.V.; Masyuk, A.I.; LaRusso, N.F. Polycystic Liver Disease: Advances in Understanding and Treatment. Annu. Rev. Pathol. 2022, 17, 251–269. [Google Scholar] [CrossRef]
- Chauveau, D.; Fakhouri, F.; Grünfeld, J.-P. Liver involvement in autosomal-dominant polycystic kidney disease: Therapeutic dilemma. J. Am. Soc. Nephrol. 2000, 11, 1767–1775. [Google Scholar] [CrossRef]
- Perrone, R.D.; Malek, A.M.; Watnick, T. Vascular complications in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Bouleti, C.; Flamant, M.; Escoubet, B.; Arnoult, F.; Milleron, O.; Vidal-Petiot, E.; Langeois, M.; Ou, P.; Vrtovsnik, F.; Jondeau, G. Risk of Ascending Aortic Aneurysm in Patients with Autosomal Dominant Polycystic Kidney Disease. Am. J. Cardiol. 2019, 123, 482–488. [Google Scholar] [CrossRef]
- Lee, T.-L.; Chen, C.-F.; Tan, A.C.; Chan, C.-H.; Ou, S.-M.; Chen, F.-Y.; Yu, K.-W.; Chen, Y.-T.; Lin, C.-C. Prognosis of Vascular Access in Haemodialysis Patients with Autosomal Dominant Polycystic Kidney Disease. Sci. Rep. 2020, 10, 1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, A.; Donfrid, B.; Adam, J.; Ilic, M.; Djuric, Z.; Damjanovic, T.; Popovic, J.; Popovic, G.; Radojicic, Z.; Dimkovic, N. Arteriovenous fistula aneurysm in patients on regular hemodialysis: Prevalence and risk factors. Nephron Clin. Pract. 2013, 124, 94–98. [Google Scholar] [CrossRef]
- Hadimeri, H.; Hadimeri, U.; Attman, P.O.; Nyberg, G. Dimensions of arteriovenous fistulas in patients with autosomal dominant polycystic kidney disease. Nephron 2000, 85, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Paavola, J.; Schliffke, S.; Rossetti, S.; Kuo, I.Y.-T.; Yuan, S.; Sun, Z.; Harris, P.C.; Torres, V.E.; Ehrlich, B.E. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 58, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Sagredo, A.; Quiroga, C.; Garrido-Moreno, V.; López-Crisosto, C.; Leiva-Navarrete, S.; Norambuena-Soto, I.; Ortiz-Quintero, J.; Díaz-Vesga, M.C.; Perez, W.; Hendrickson, T.; et al. Polycystin-1 regulates cardiomyocyte mitophagy. FASEB J. 2021, 35, e21796. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, B.A.; Kotaro, Y.; Martin, W.; Sharma, A.; Kamath, P.S.; Edwards, M.E.; Kremers, W.K.; Chari, S.T.; Torres, V.E.; Harris, P.C.; et al. Pancreatic Cysts and Intraductal Papillary Mucinous Neoplasm in Autosomal Dominant Polycystic Kidney Disease. Pancreas 2019, 48, 698–705. [Google Scholar] [CrossRef]
- Torra, R.; Sarquella, J.; Calabia, J.; Martí, J.; Ars, E.; Fernández-Llama, P.; Ballarin, J. Prevalence of cysts in seminal tract and abnormal semen parameters in patients with autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 790–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, C.K.; Zeligman, B.E.; Johnson, A.M.; Duley, I.; Gabow, P.A. Evaluation of colonic diverticular disease in autosomal dominant polycystic kidney disease without end-stage renal disease. Am. J. Kidney Dis. 1999, 34, 863–868. [Google Scholar] [CrossRef]
- Kumar, S.; Adeva, M.; King, B.F.; Kamath, P.S.; Torres, V.E. Duodenal diverticulosis in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2006, 21, 3576–3578. [Google Scholar] [CrossRef] [Green Version]
- Morris-Stiff, G.; Coles, G.; Moore, R.; Jurewicz, A.; Lord, R. Abdominal wall hernia in autosomal dominant polycystic kidney disease. Br. J. Surg. 1997, 84, 615–617. [Google Scholar]
- Modi, K.B.; Grant, A.C.; Garret, A.; Rodger, R.S. Indirect inguinal hernia in CAPD patients with polycystic kidney disease. Adv. Perit. Dial. 1989, 5, 84–86. [Google Scholar] [PubMed]
- Dupont, V.; Kanagaratnam, L.; Sigogne, M.; Bechade, C.; Lobbedez, T.; Portoles, J.; Rieu, P.; Drame, M.; Touré, F. Outcome of polycystic kidney disease patients on peritoneal dialysis: Systematic review of literature and meta-analysis. PLoS ONE 2018, 13, e0196769. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.; Iaria, G.; Pellicciaro, M.; Sforza, D.; Parente, A.; Campisi, A.; Cacciatore, C.; Calafiore, E.; Pisani, G.; Tisone, G. Native Nephrectomy in Patients with Autosomal Dominant Polycystic Kidney Disease Evaluated for Kidney Transplantation. Transplant. Proc. 2019, 51, 2914–2916. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, P.K.; Vuento, M.; Maunola, M.; Ala-Houhala, I. Ovarian manifestations in women with autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 2002, 40, 504–507. [Google Scholar] [CrossRef]
- Moua, T.; Zand, L.; Hartman, R.P.; Hartman, T.E.; Qin, D.; Peikert, T.; Qian, Q. Radiologic and clinical bronchiectasis associated with autosomal dominant polycystic kidney disease. PLoS ONE 2014, 9, e93674. [Google Scholar] [CrossRef] [Green Version]
- Viggiano, D.; Bruchfeld, A.; Carriazo, S.; De Donato, A.; Endlich, N.; Ferreira, C.; Figurek, A.; Fouque, D.; Franssen, C.F.M.; Giannakou, K.; et al. Brain dysfunction in tubular and tubulointerstitial kidney diseases. Nephrol. Dial. Transplant. 2021, 37 (Suppl. S2), ii46–ii55. [Google Scholar] [CrossRef]
- Viggiano, D.; Wagner, C.A.; Blankestijn, P.J.; Bruchfeld, A.; Fliser, D.; Fouque, D.; Frische, S.; Gesualdo, L.; Gutiérrez, E.; Goumenos, D.; et al. Mild cognitive impairment and kidney disease: Clinical aspects. Nephrol. Dial. Transplant 2020, 35, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, D.; Wagner, C.A.; Martino, G.; Nedergaard, M.; Zoccali, C.; Unwin, R.; Capasso, G. Mechanisms of cognitive dysfunction in CKD. Nat. Rev. Nephrol. 2020, 16, 452–469. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Ortiz, A.; Blumbyte, I.A.; Rudolf, S.; Beck-Sickinger, A.G.; Malyszko, J.; Spasovski, G.; Carriazo, S.; Viggiano, D.; Kurganaite, J.; et al. Neuropeptide Y as a risk factor for cardiorenal disease and cognitive dysfunction in CKD: Translational opportunities and challenges. Nephrol. Dial. Transplant. 2021, 37 (Suppl. S2), ii14–ii23. [Google Scholar] [CrossRef]
- Liabeuf, S.; Pepin, M.; Franssen, C.F.M.; Viggiano, D.; Carriazo, S.; Gansevoort, R.T.; Gesualdo, L.; Hafez, G.; Malyszko, J.; Mayer, C.; et al. Chronic kidney disease and neurological disorders: Are uraemic toxins the missing piece of the puzzle? Nephrol. Dial. Transplant. 2021, 37 (Suppl. S2), ii33–ii44. [Google Scholar] [CrossRef]
- Bikbov, B.; Soler, M.J.; Pešić, V.; Capasso, G.; Unwin, R.; Endres, M.; Remuzzi, G.; Perico, N.; Gansevoort, R.; Mattace-Raso, F.; et al. Albuminuria as a risk factor for mild cognitive impairment and dementia—What is the evidence? Nephrol. Dial. Transplant. 2021, 37 (Suppl. S2), ii55–ii62. [Google Scholar] [CrossRef] [PubMed]
- Imenez Silva, P.H.; Unwin, R.; Hoorn, E.J.; Ortiz, A.; Trepiccione, F.; Nielsen, R.; Pesic, V.; Hafez, G.; Fouque, D.; Massy, Z.A.; et al. Acidosis, cognitive dysfunction and motor impairments in patients with kidney disease. Nephrol. Dial. Transplant. 2021, 37 (Suppl. S2), ii4–ii12. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, D. The hyperactive syndrome: Metanalysis of genetic alterations, pharmacological treatments and brain lesions which increase locomotor activity. Behav. Brain Res. 2008, 194, 1–14. [Google Scholar] [CrossRef]
- Viggiano, A.; Cacciola, G.; Widmer, D.A.J.D.; Viggiano, D. Anxiety as a neurodevelopmental disorder in a neuronal subpopulation: Evidence from gene expression data. Psychiatry Res. 2015, 228, 729–740. [Google Scholar] [CrossRef]
- De Sanctis, C.; Bellenchi, G.C.; Viggiano, D. A meta-analytic approach to genes that are associated with impaired and elevated spatial memory performance. Psychiatry Res. 2018, 261, 508–516. [Google Scholar] [CrossRef]
- Kurtul, B.E.; Elbeyli, A.; Kakac, A.; Turgut, F. Corneal endothelial cell density and microvascular changes of retina and optic disc in autosomal dominant polycystic kidney disease. Indian J. Ophthalmol. 2021, 69, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, R.A.; Nagaraju, S.P.; Addoor, K.R. Retinal detachment in autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 2014, 63, 541–542. [Google Scholar] [CrossRef] [PubMed]
- dell’Omo, R.; Scupola, A.; Viggiano, D.; Sammarco, G.M.; De Turris, S.; Romano, M.M.R.M.; Grimaldi, G.; dell’Omo, E.; Costagliola, C.; Sammarco, M.G.; et al. Incidence and factors influencing retinal displacement in eyes treated for rhegmatogenous retinal detachment with vitrectomy and gas or silicone oil. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO191–BIO199. [Google Scholar] [CrossRef]
- Dell’Omo, R.; Viggiano, D.; Giorgio, D.; Filippelli, M.; Di Iorio, R.; Calo, R.; Cardone, M.; Rinaldi, M.; Omo, E.; Costagliola, C.; et al. Restoration of foveal thickness and architecture after macula-off retinal detachment repair. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.; Pyeritz, R.E.; Litt, H.I. Abdominal visceral findings in patients with Marfan syndrome. Genet. Med. 2007, 9, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wang, C.J.; Judge, D.P.; Halushka, M.K.; Ni, J.; Habashi, J.P.; Moslehi, J.; Bedja, D.; Gabrielson, K.L.; Xu, H.; et al. A Pkd1-Fbn1 genetic interaction implicates TGF-β signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.M.; Sikaneta, T.; Sullivan, B.M.; Zhang, Q.; Andreucci, M.; Stehle, T.; Drummond, I.; Arnaout, M.A. Polycystin-1 interacts with intermediate filaments. J. Biol. Chem. 2001, 276, 46544–46552. [Google Scholar] [CrossRef] [Green Version]
- Scheffers, M.S.; van der Bent, P.; Prins, F.; Spruit, L.; Breuning, M.H.; Litvinov, S.V.; de Heer, E.; Peters, D.J. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum. Mol. Genet. 2000, 9, 2743–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignogna, P.; Viggiano, D. Brain distribution of genes related to changes in locomotor activity. Physiol. Behav. 2010, 99, 618–626. [Google Scholar] [CrossRef]
- Qian, F.; Watnick, T.J.; Onuchic, L.F.; Germino, G.G. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996, 87, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.Y.; Zhang, T.; Michaeel, A.; Blumenfeld, J.; Liu, G.; Zhang, W.; Zhang, Z.; Zhu, Y.; Rennert, L.; Martin, C.; et al. Somatic Mutations in Renal Cyst Epithelium in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2139–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grantham, J.J. The etiology, pathogenesis, and treatment of autosomal dominant polycystic kidney disease: Recent advances. Am. J. Kidney Dis. 1996, 28, 788–803. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.J.; Eacobacci, K.; Goyal, R. The Incidental Renal Mass- Update on Characterization and Management. Radiol. Clin. N. Am. 2021, 59, 631–646. [Google Scholar] [CrossRef]
- Schwarz, A.; Vatandaslar, S.; Merkel, S.; Haller, H. Renal cell carcinoma in transplant recipients with acquired cystic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lim, J.H.; Jeong, I.G.; Choe, J.; Kim, C.-S.; Hong, J.H. What association exists between hypertension and simple renal cyst in a screened population? J. Hum. Hypertens. 2013, 27, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Xiao, Y.; Xiong, X.; Li, L.; Yang, Y.; Han, Y.; Zhao, H.; Yang, M.; Sun, L. The Relationship Between Simple Renal Cysts and Renal Function in Patients with Type 2 Diabetes. Front. Physiol. 2020, 11, 616167. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Diameter (µm) | Intratubular Pressure (mmHg) | Wall Tension (τ) | |
|---|---|---|---|
| Proximal tubule | 23 | 8.1 | 0.054 |
| Loop of Henle | 15 | 4 | 0.027 |
| Distal convoluted tubule | 40 | 4 | 0.027 |
| Collecting duct | 24 | 2 | 0.013 |
| Tubular Tract | % of Occurrence |
|---|---|
| Proximal tubule | 1.8–5% |
| Loop of Henle | Not available |
| Distal convoluted tubule | 42% |
| Collecting duct | 7–40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Corte, M.; Viggiano, D. Wall Tension and Tubular Resistance in Kidney Cystic Conditions. Biomedicines 2023, 11, 1750. https://doi.org/10.3390/biomedicines11061750
Della Corte M, Viggiano D. Wall Tension and Tubular Resistance in Kidney Cystic Conditions. Biomedicines. 2023; 11(6):1750. https://doi.org/10.3390/biomedicines11061750
Chicago/Turabian StyleDella Corte, Michele, and Davide Viggiano. 2023. "Wall Tension and Tubular Resistance in Kidney Cystic Conditions" Biomedicines 11, no. 6: 1750. https://doi.org/10.3390/biomedicines11061750
APA StyleDella Corte, M., & Viggiano, D. (2023). Wall Tension and Tubular Resistance in Kidney Cystic Conditions. Biomedicines, 11(6), 1750. https://doi.org/10.3390/biomedicines11061750