
Genetic, Phenotypic, and Clinical Heterogeneity of NPM1-Mutant Acute Myeloid Leukemias


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
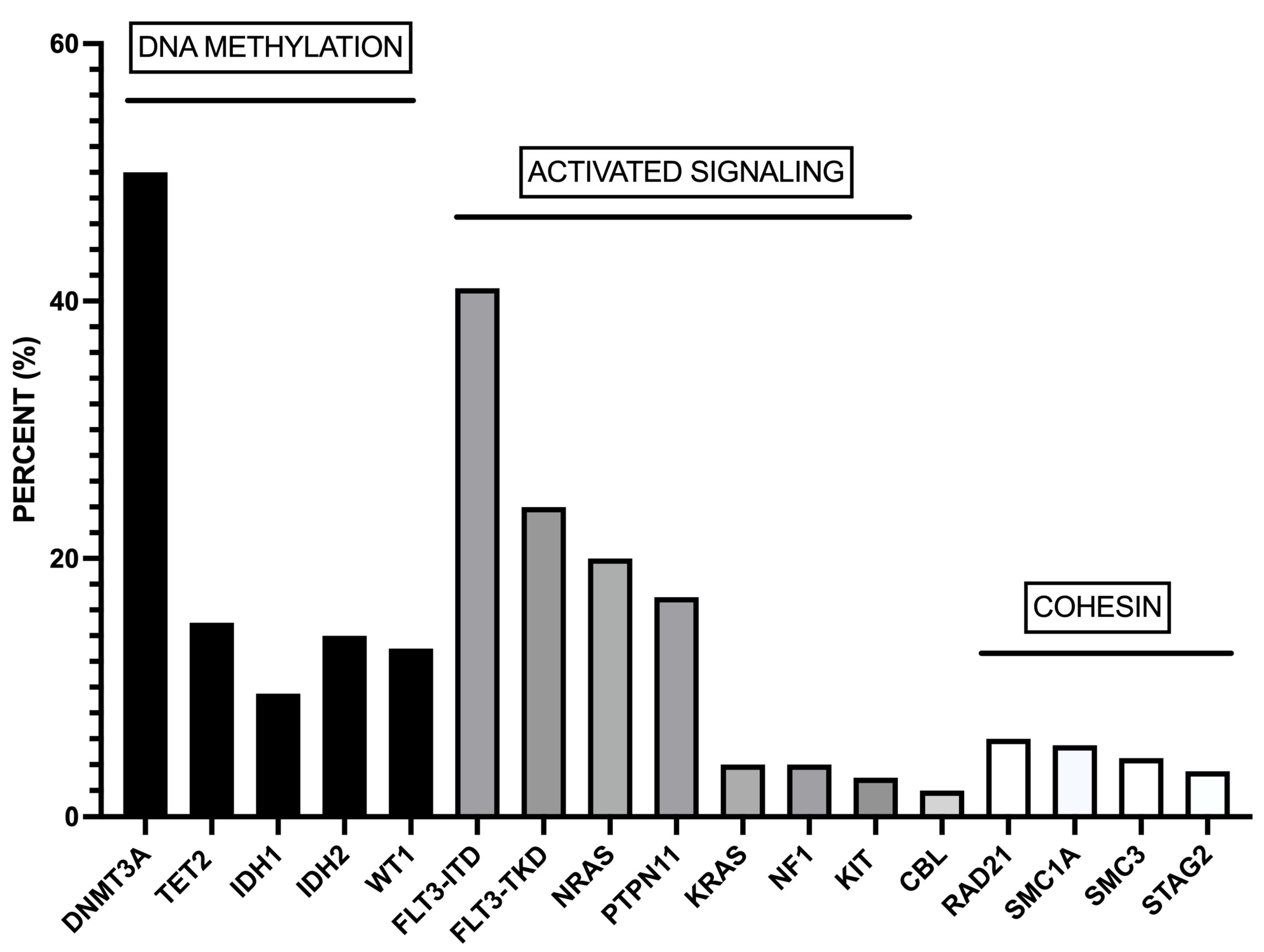
2. The Mutational Landscape of NPM1-Mutant AMLs
3. Cell Differentiation Heterogeneity of NPM1-Mut AMLs
4. Clonal Architecture and Clonal Evolution of NPM1-Mutant AMLs
5. Prognostic Heterogeneity of NPM1-Mut AMLs
6. DNMT3A Mutations in NPM1-Mut AMLs
7. FLT3 Mutations in NPM1-mut AML Patients
8. IDH1 and IDH2 Mutations in NPM1-Mut AMLs
9. Cohesin Complex Gene Mutations in NPM1-mut AMLs
10. RAS Mutations in NPM1-Mut AMLs
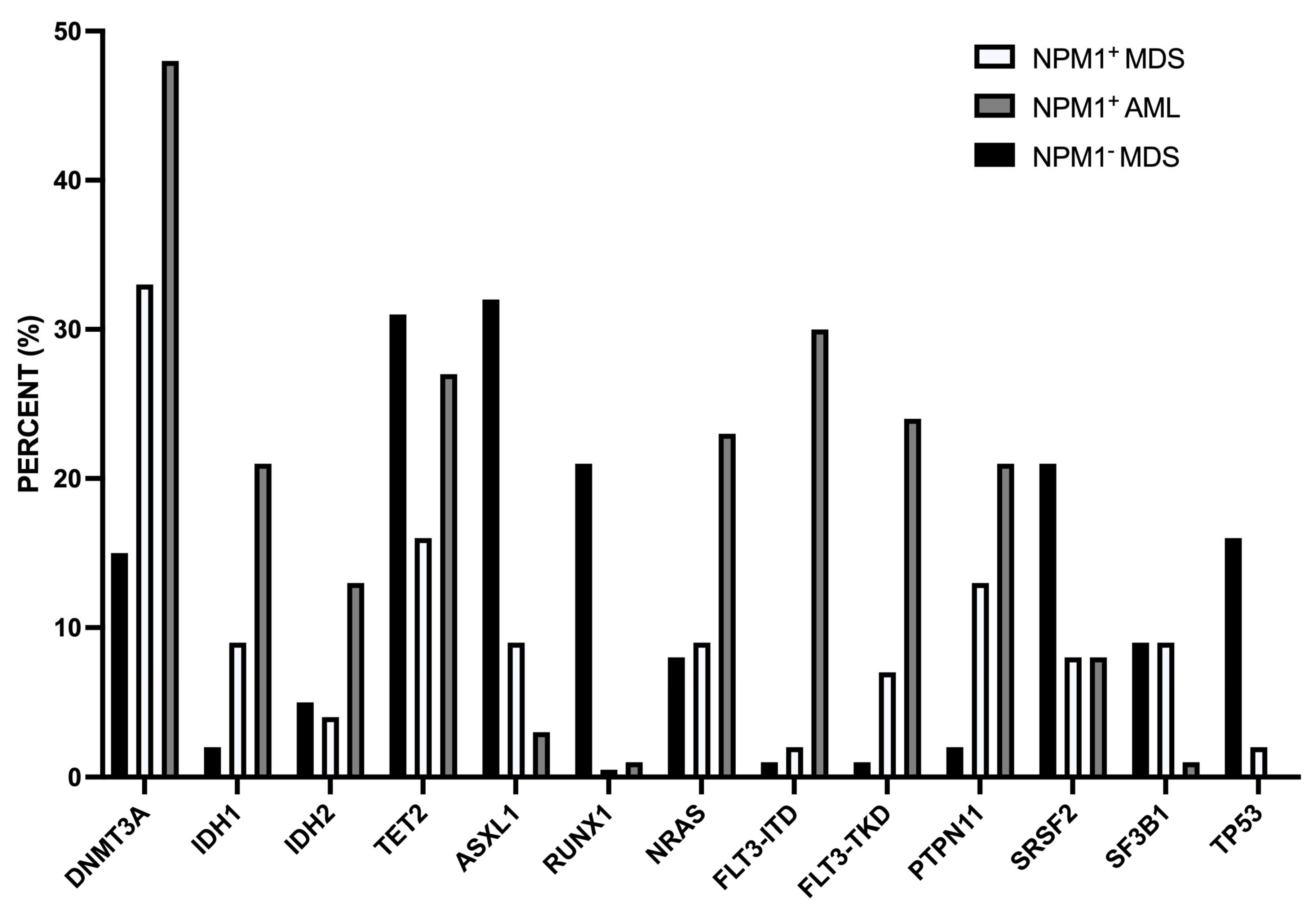
11. Myelodysplasia-Related Alterations in NPM1-Mut AMLs
11.1. NPM1-Mut MDS
11.2. NPM1-Mut AMLs with MDS-Related Mutations
11.3. NPM1-Mut Secondary AMLs
12. TET2 Mutations in IDH1-Mut AML
13. PTPN11 Mutations in NPM1-Mut AMLs
14. Therapy of NPM1-Mut AMLs
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, K.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Hauser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk stratification in acute myeloid leukemia. Nat. Commun. 2022, 13, 4622. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of myeloid neoplasms and acute leukemia: Integrating morphological, clinical and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla OAkkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan John, K.C.; Chen, W.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1720. [Google Scholar] [CrossRef]
- Angenendt, L.; Rollig, C.; Montesinos, P.; Martinez-Cuadròn, D.; Barragan, E.; Garcia, R.; Botella, C.; Martinez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal abnormalities and prognosis in NPM1-mutated acute myeloid leukemia: A pooled analysis of individual patient data from nine international cohorts. J. Clin. Oncol. 2019, 37, 2632–2642. [Google Scholar] [CrossRef]
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.M.; et al. Molecular subtypes of NPM1 mutations have different clinical profiles specific patterns of accompanying molecular mutations and varying outcome in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, e55. [Google Scholar] [CrossRef] [Green Version]
- Bailey, G.D.; Doolan, L.; Baskar, A.; Smith, L.C.; Seedhouse, C.H. Preferential transcription of the mutated allele in NPM1 mutated acute myeloid leukemia. Sci. Rep. 2020, 10, 17695. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.S.; Ramabradan, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckelmann, H.J.; Haarer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; Perner, F.; Rajput, M.; Antonissen, N.; Wen, Y.; et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov. 2023, 13, 746–765. [Google Scholar] [CrossRef]
- Wang, X.Q.D.; Fan, D.; Han, Q.; Liu, Y.; Miao, H.; Wang, X.; Li, Q.; Chen, D.; Gore, H.; Himadewi, P.; et al. Mutant NPM1 hijacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov. 2023, 13, 724–745. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, A.; Ammer, T.; Kechter, A.; Rawat, V.; Sinha, A.; Gonzalez-Menendiez, I.; Quintanilla-Martinez, L.; Azoitein, A.; Gunes, C.; Mupo, A.; et al. Npm1 haploinsufficiency in collaboration with MEIS1 is sufficient to induce AML in mice. Blood Adv. 2023, 7, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, M.A.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard-risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Sargas, C.; Ayala, R.; Larrayoz, M.J.; Chillon, M.C.; Carillo-Cruz, E.; Bialbao-Sieyro, C.; Prados de la Torre, E.; Martinez-Cadron, D.; Rodriguez-Veiga, R.; Boluda, B.; et al. Molecular landscape and validation of new genomic classification in 2668 adult AML patients: Real life data from the PETHEMA group. Cancers 2023, 15, 438. [Google Scholar] [CrossRef]
- Petterson, L.; Holmgren, B.; Juliusson, G.; Lazaveric, V.; Ehinger, M. Mutational spectrum of de novo NPM1-mutated acute myeloid leukemia patients older than 75 years. Leuk Lymphoma 2021, 62, 1958–1966. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Outcomes of older patients with NPM1-mutated AML: Current treatments and the promise of venetoclax-based regimens. Blood Adv. 2020, 4, 1311–1320. [Google Scholar] [CrossRef]
- Khan, A.M.; Reddy, S.N.; Aly, M.; Dhillon, V.; Sbihi, A.A.; Kewan, T.; Bahaj, W.; Gurnari, C.; Al_Share, B.; Dyson, G.; et al. Comprehensive age-stratified impact of NPM1-Mutation in acute myeloid leukemia. Blood 2022, 140, 1433–1434. [Google Scholar] [CrossRef]
- Cantu, M.D.; Kanagal-Shamanna, R.; Wang, S.; Kadia, T.; Bueso-Ramos, C.E.; Patel, S.S.; Geyer, T.T.; Tam, W.; Madanat, Y.; Li, P.; et al. Clinicopathologic and molecular analysis of normal karyotype-related and de novo acute myeloid leukemia: A multi-institutional study by the bone marrow pathology group. JCO Precis. Oncol. 2023, 7, e2200400. [Google Scholar] [CrossRef] [PubMed]
- Ohtman, J.; Meggendorfer, M.; Tiacci, E.; Thiede, C.; Schlenk, R.; Dillon, R.; Stasik, S.; Venanzi, A.; Bertoli, S.; Delabesse, E.; et al. Overlapping features of therapy-related and de novo NPM1-mutated AML. Blood 2023, 141, 1846–1857. [Google Scholar]
- Mason, E.F.; Hasserjian, R.P.; Aggarwal, N.; Seegmiller, A.C.; Podznyakova, O. Blast phenotype and comutations in acute myeloid leukemia with mutated NPM1 influence disease biology and outcome. Blood Adv. 2019, 3, 3322–3332. [Google Scholar] [CrossRef] [Green Version]
- Mer, A.S.; Haeth, E.M.; Tonekaboni, S.A.M.; Dogan-Artun, N.; Nair, S.K.; Murison, A.; Garcia-Prat, L.; Shlush, L.; Hurren, R.; Voisin, V.; et al. Biological and therapeutic implications of a unique subtype pf NPM1 mutated AML. Nat. Commun. 2021, 12, 1054. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Li, J.F.; Zhu, Y.M.; Lin, X.J.; Wen, L.J.; Zhang, F.; Zhang, Y.L.; Zhao, M.; Fang, H.; Wang, S.Y.; et al. Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2022, 119, e2211429119. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Kern, W.; Haferlach, T.; Haferlach, C.; Hollein, A. NPM1 mutated AML is characterized by pre-leukemic mutations and the persistence and acquisition of co-mutations in molecular remission leads to inferior prognosis. Blood 2018, 132 (Suppl. 1), 996. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs. CHOP mutations in AML with NPM1 alteration. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Graeves, M. Single cell analysis of clonal architecture in acute myeloid leukemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Marcia-Trichant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Ploen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br. J. Haematol. 2014, 167, 478–486. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Weber, D.; Paschka, P.; Kaumanns, A.; Krieger, S.; Carbacioglu, A.; Kronke, J.; Kapp-Schworer, S.; Kramer, D.; Horst, H.A.; et al. DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia 2018, 32, 30–37. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Kronke, J.; Bullinger, L.; Teleanu, V.; Tschrtz, F.; Gaidzik, V.I.; Kuhn, M.; Rucker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schworer, S.; et al. Clonal evolution in relapsed NPM1-mutatted acute myeloid leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Hollein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef]
- Cocciardi, S.; Dolnik, A.; Kapp-Schoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Sanchez, A.; Villaverde-Ramiro, A.; Elicegui, J.M.; Gonzalez, T.; Benner, A.; Strong, E.; Castellani, G.; Eckman, C.A.; Versluis, J.; Abaigar, M.; et al. NPM1 mutated AML: Impact of co-mutational patterns. Results of the European HARMONY Alliance. HemaSphere 2022, 6, S3. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, A.; Villaverde-Ramiro, A.; Strang, E.; Castellani, G.; Heckman, C.A.; Versluis, J.; Abaigar, M.; Sobas, M.A.; Melchor, R.A.; Benner, A.; et al. Machine learning allows the identification on new co-mutational patterns with prognostic implications in NPM1-mutated AML: Results of the European Hasrmiony Alliance. Blood 2022, 140 (Suppl. 1), 739–742. [Google Scholar] [CrossRef]
- Mrozek, K.; Kohlschmidt, J.; Blachly, J.S.; Nicolet, D.; Carroll, A.J.; Archer, K.J.; Mims, A.S.; Lerkin, K.T.; Orwick, S.; Oakes, C.C.; et al. Outcome prediction by the 2022 European Leukemia Net genetic-risk classification for adults with acute myeloid leukemia: An Alliance study. Leukemia 2023, 37, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Rollig, C.; Montesinos, P.; Ravandi, F.; Juliusson, G.; Récher, C.; Htzykson, R.; Racil, Z.; Wei, A.H.; Schliemann, C. Revisiting coexisting chromosomal abnormalities in NPM1-mutated AML in light of the revised ELN 2022 classification. Blood 2023, 141, 433–435. [Google Scholar] [CrossRef]
- Patel, S.S.; Kuo, F.C.; Gibson, C.J.; Steensma, D.P.; Soiffer, R.J.; Edwin, A., III.; Chen, Y.B.A.; Fathi, A.T.; Graubert, T.A.; Brunner, A.M.; et al. High NPM1-mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood J. Am. Soc. Hematol. 2018, 131, 2816–2825. [Google Scholar] [CrossRef] [Green Version]
- Abbas, H.A.; Ravandi, F.; Loghavi, S.; Borthakur, G.; Kadia, T.M.; Jabbour, E.; Takahashi, K.; Cortes, J.; Issa, G.C.; Konopleva, M.; et al. NPM1 mutant variant allele frequency correlates with leukemia burden but does not provide prognostic information in NPM1-mutated AML. Am. J. Hematol. 2019, 94, E158–E160. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg-Thurley, M.; Herold, T.; Gorlich, D.; Sauerland, C.; Janke, H.; Prassek, V.V.; Konstandin, N.P.; Dufour, A.M.; Schneider, S.; Ksienzyk, B.; et al. NPM1 variant allele frequency and outcomes in AML. Blood 2018, 132 (Suppl. 1), 1486. [Google Scholar] [CrossRef]
- Patel, S.S.; Pinkus, G.S.; Ritterhouse, L.L.; Segal, J.P.; Dal Cin, P.; Restrepo, T.; Harris, M.H.; Stone, R.M.; Hasserjian, R.P.; Weinberg, O.K. High NPM1 mutant allele burden at diagnosis correlates with minimal residual disease at first remission in de novo acute myeloid leukemia. Am. J. Hematol. 2019, 94, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.V.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Hutter, S.; Kern, W.; Haferlach, T.; Haferlach, C.; Hollen, A. DNMT3A mutations are over-represented in young adults with NPM1 mutated AML and prompt a distinct co-mutational pattern. Leukemia 2019, 33, 2741–2746. [Google Scholar] [CrossRef]
- Chin, L.; Wong, C.Y.G.; Gill, H. Targeting and monitoring acute myeloid leukemia with nucleophosmin-1 (NPM1) mutation. Int. J. Mol. Sci. 2023, 24, 3161. [Google Scholar] [CrossRef] [PubMed]
- Onate, G.; Bataller, A.; Garrido, A.; Hoyos, M.; Vives, S.; Coll, R.; Tommo, M.; Sampol, A.; Escoda, L.; Salamero, O.; et al. Prognostic impact of DNMT3A mutation in acute myeloid leukemia with mutataed NPM1. Blood Adv. 2022, 6, 882–890. [Google Scholar] [CrossRef]
- Loghavi, S.; Zuo, Z.; Ravandi, F.; Kantajian, H.M.; Bueso-Ramos, C.; Zhang, L.; Singh, R.R.; Patel, K.P.; Medeiros, L.J.; Stingo, F.; et al. Clinical features of de novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J. Hematol. Oncol. 2014, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Reyes-Palomares, A.; He, L.; Bergeron, A.; Lavallée, V.P.; Lemieux, S.; Gendron, P.; Rohde, C.; Xia, J.; Jaghdane, P.; et al. Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3-ITD triple-mutatedAML. Blood 2019, 134, 263–276. [Google Scholar] [CrossRef]
- Beserra, M.F.; Lima, A.S.; Piqué-Borras, M.R.; Silveira, D.R.; Coelho-Silva, J.L.; Pereira-Martins, D.A.; Weinhauser, I.; Franca-Neto, P.L.; Quek, L.; Corby, A.; et al. Co-occurrence of DNMT3A, NPM1, FLT3 mutations identifies a subset of acute myeloid leukemia with adverse prognosis. Blood 2020, 135, 870–875. [Google Scholar] [CrossRef]
- Holz-Scheitinger, C.; Matie, D.M.; Reich, N.O. Mutations in Dna methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Kahler, A.K.; Hnadsaker, R.E. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequences. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Wakita, S.; Marumo, A.; Morita, K.; Kako, S.; Toya, T.; Najima, Y.; Doki, N.; Kanda, J.; Kuroda, J.; Mori, S.; et al. Mutational analysis on DNMT3A improves the prognostic stratification of patients with acute myeloid leukemia. Cancer Sci. 2013, 114, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Rucker, F.G.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; Appelbaum, F.R.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2022, 36, 90–99. [Google Scholar] [CrossRef]
- Chan, O.; Al Ali, N.; Ball, S.; Grenet, J.; Hana, C.; Deutsch, Y.F.; Tashkandi, H.; Zhang, L.; Hussaini, M.O.; Yun, S.; et al. The prognostic impact of FLT3 in NPM1-mutated AML: Co-occurrence of FLT3-ITD and FLT3-TKD confers poor outcomes. Blood 2022, 140 (Suppl. 1), 3435–3437. [Google Scholar] [CrossRef]
- Jentsch, M.; Bischof, L.; Brauer, D.; Backaus, D.; Ussmann, J.; Franke, G.N.; Vucinic, V.; Platzbecker, U.; Schwind, S. Clinical implications of the FLT3-ITD allelic ratio in acute myeloid leukemia in the context of an allogeneic stem cell transplantation. Cancers 2023, 15, 1312. [Google Scholar] [CrossRef]
- Grob, T.; Sanders, M.A.; Vonk, C.M.; Kavelaars, F.G.; Rijken, M.; Hanekamp, D.; Gradowska, P.L.; Cloos, J.; Floisand, Y.; Kooy, M.M.; et al. Prognostic value of FLT3-internal tandem duplication residual disease in acute myeloid leukemia. J. Clin. Oncol. 2023, 41, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Middeke, J.M.; Metzeler, K.H.; Rollig, C.; Kramer, M.; Eckardt, J.N.; Stasik, S.; Greif, P.A.; Spiekermann, K.; Rothenbrg-Thurley, M.; Krug, U.; et al. Differential impact of IDH1/2 mutational subclasses on outcome in adult AML: Results from a large munlticenter study. Blood Adv. 2022, 6, 1394–1403. [Google Scholar] [CrossRef]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. IDH1-R132 changes vary according to NPM1 and other mutations status in AML. Leukemia 2019, 33, 1043–1047. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Cappelli, L.V.; Walter, W.; Haferlach, C.; Kern, W.; Falini, B.; Haferlach, T. IDH1R132, IDH2R140 and IDH2R172 in AML: Different genetic landscapes correlate with outcome and may influence targeted treatment strategies. Leukemia 2018, 32, 1249–1253. [Google Scholar] [CrossRef]
- Eckardt, J.N.; Stasik, S.; Rollig, C.; Sauer, T.; Scholl, S.; Hochaus, A.; Crysandt, M.; Bummendorf, T.; Naumann, R.; Steffen, B.; et al. Alterations of cohesin complex genes in acute myeloid leukemia: Differential co-mutations, clinical presentation and impact on outcome. Blood Cancer J. 2023, 13, 18. [Google Scholar] [CrossRef]
- Simonetti, G.; Mengucci, C.; Padella, A.; Fonzi, E.; Picone, G.; Delpino, C.; Nanni, J.; De Tommaso, R.; Franchini, E.; Papayannidis, C.; et al. Integrated genomic-metabolic classification of acute myeloid leukemia defines a subgroup with NPM1 and cohesin/DNA damage mutations. Leukemia 2021, 35, 2813–2826. [Google Scholar] [CrossRef]
- Benard, B.A.; Leak, L.B.; Azizi, A.; Thomas, D.; Gentles, A.J.; Majeti, R. Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nat. Commun. 2021, 12, 7244. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.E.; Stelloh, C.; Pulakanti, K.; Burns, R.; Fisher, J.B.; Heimbruch, K.E.; Tarima, S.; Furumo, Q.; Brennan, J.; Zheng, Y.; et al. Cominatorial genetics reveals the Dock1-Rac2 axis as a potential target for the treatment of NPM1; Cohesin mutated AML. Leukemia 2022, 36, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Z.; Li, T.; Li, Y.; Wang, W.; Hao, Q.; Xie, X.; Wan, D.; Jiang, Z.; Wang, C.; et al. Mutational sepctrum and prognosis in NRAS-mutated acute myeloid leukemia. Sci. Rep. 2020, 10, 12152. [Google Scholar] [CrossRef] [PubMed]
- Rivera, D.; Kim, K.; Kanagal-Shamanna, R.; Borthakur, G.; Montalban-Bravo, G.; Daver, N.; DiNardo, C.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; et al. Implications of RAS mutational status in subsets of patients with newly diagnosed acute myeloid leukemia across therapy subtypes. Am. J. Hematol. 2022, 97, 1599–1606. [Google Scholar] [CrossRef]
- Arber, D.A.; Erba, H.P. Diagnosis and treatment of patients with acute myeloid leukemia with myelodysplastic changes (AML-MRC). Am. J. Clin. Pathol. 2020, 154, 731–741. [Google Scholar] [CrossRef]
- Bains, A.; Luthra, R.; Medeiros, L.J.; Zuo, Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: Frequency and potential value for predicting progression to acute myeloid leukemia. Am. J. Clin. Pathol. 2011, 135, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [Green Version]
- Forghieri, F.; Paolini, A.; Morselli, M.; Bigliardi, S.; Bonacorsi, G.; Leonardi, G.; Coluccio, V.; Maccaferri, M.; Fantuzzi, V.; Faglioni, L.; et al. NPM1 mutations may reveal acute myeloid leukemia in cases otherwise morphologically diagnosed as myelodysplastic syndromes or myelodysplatic/myeloproliferative neoplasms. Leuk. Lymphoma 2015, 56, 3222–3226. [Google Scholar] [CrossRef]
- Maurya, N.; Mohanty, P.; Dhangar, S.; Panchal, P.; Jijina, F.; Mathan, L.P.; Shanmukhaiah, C.; Madkaikar, M.; Vudinti, B.R. Comprehensive analysis of genetic factors predicting overall survival in myelodysplastic syndromes. Sci. Rep. 2022, 12, 5925. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Ganan-Gomez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; DiNardo, C.; Borthakur, G.; et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019, 3, 922–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chan, O.; Ali, A.; Kuykendall, A.; Padron, E.; Sweet, K.; Lancet, J.; Komrokji, R.S.; Sallam, D.A. Clinical characterization and outcomes of patients with NPM1-mutated myelodysplastic syndromes or chronic myelomonocytic leukemia. Blood 2022, 140, 6936–6937. [Google Scholar] [CrossRef]
- Patel, S.S.; Ho, C.; Ptashkin, R.N.; Sadigh, S.; Bagg, A.; Geyer, J.T.; Xu, M.L.; Prebet, T.; Mason, E.F.; Seegmiller, A.C.; et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv. 2019, 3, 1540–1545. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Martelli, M.P.; Brunetti, L.; Gjerten, B.T.; Andresen, V. The NPM1 defines AML irrespective of blasts count. Am. J. Hematol. 2023, in press. [Google Scholar]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardin, C.; Pautas, C.; Fournier, E.; Itzykson, R.; Lemasle, E.; Bourhis, J.H.; Adès, L.; Marolleau, J.P.; Malfuson, J.V.; Gastaud, L.; et al. Added prognostic value of secondary AML-like gene mutations in ELN intermediate-risk older AML: ALFA-1200 study results. Blood Adv. 2020, 4, 1942–1949. [Google Scholar] [CrossRef]
- Fuhrmann, I.; Lenk, M.; Haferlach, T.; Stengel, A.; Hutter, S.; Baer, C.; Meggendorfer, M.; Kern, W.; Haferlach, C. AML, NOS and AML-MRC as defined by multilineage dysplasia share a common mutation pattern which is distinct from AML-MRC as defiend by MDS-related cytogenetics. Leukemia 2022, 36, 1929–1942. [Google Scholar] [CrossRef]
- Tsai, X.; Sun, K.J.; Lo, N.Y.; Tien, F.M.; Kuo, Y.Y.; Tseng, M.H.; Peng, Y.L.; Chuang, Y.K.; Ko, B.S.; Tang, J.L.; et al. Poor prognostic implications of myelodysplastic-related mutations in both older and younger patients with de novo AML. Blood Cancer J. 2023, 13, 4. [Google Scholar] [CrossRef]
- Chan, O.; Ali, N.A.; Tashkandi, H.; Ellis, A.; Ball, S.; Grenet, J.; Hana, C.; Deutsch, Y.E.; Zhang, L.; Hussaini, M.O.; et al. Secondary AML mutations confer poor prognosis in patients with ELN favorable risk NPPM1-mutated AML. Blood 2022, 140 (Suppl. 1), 3419–3421. [Google Scholar] [CrossRef]
- Wang, Y.; Quesada, A.E.; Zuo, Z.; Medeiros, L.J.; Yin, C.C.; Li, S.; Xu, J.; Borthakur, G.; Li, Y.; Yang, C.; et al. The impact of mutation of myelodysplasia-related genes in de novo acute myeloid leukemia carrying NPM1 mutation. Cancers 2023, 15, 198. [Google Scholar] [CrossRef]
- Smith, E.C.; Atenafu, E.G.; Bankar, A.; Chan, S.M.; Davidson, M.B.; Gupta, V.; Minden, M.D.; Richard-Carpentier, G.; Schimmer, A.D.; Schuh, A.C.; et al. Clinical outcomes in de novo versus secondary NPM1-mutated AML. Blood 2022, 140 (Suppl. 1), 8952–8953. [Google Scholar] [CrossRef]
- Zhao, D.; Zairf, M.; Eladl, E.; Capo-Chichi, J.M.; Smith, A.C.; Atenafu, E.G.; Tierens, A.; Minden, M.D.; Schuh, A.; Chang, H. NPM1-mutated AML-MRC diagnosed on the basis of history of MDS or MDS/MPN frequently harbours secondary-type mutations and confers inferior outcome compared to AML with mutated NPM1. Leuk. Res. 2022, 118, 106869. [Google Scholar] [CrossRef] [PubMed]
- Tien, F.M.; Hou, H.A.; Tsai, C.T.; Tang, J.L.; Chen, C.Y.; Kuo, Y.Y.; Li, C.C.; Lin, C.T.; Yao, M.; Huang, S.Y. Hyperleukocytosis is associated with distinct genetic alterations and is an independent poor-risk factor in de novo acute myeloid leukemia patients. Eur. J. Haematol. 2018, 101, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Pastore, F.; Pastore, A.; Rothenberg-Thurley, M.; Metzler, K.H.; Ksienzyk, B.; Schneider, S.; Bohlander, S.K.; Braess, J.; Sauerland, M.C.; Gorlich, D.; et al. Molecular profiling of patients with cytogenetically normal acute myeloid leukemia and hyperleukocytosis. Cancer 2022, 128, 4213–4222. [Google Scholar] [CrossRef]
- Ramdas, B.; Reddy, P.L.; Mali, R.S.; Pasupuleti, S.K.; Zhang, J.; Kelley, M.R.; Paczesny, S.; Zhang, C.; Kapur, R. Combined heterozygosity of FLT3ITD, TET2, and DNMT3A results in aggressive leukemia. JCI Insight 2022, 7, e162016. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Xu, Y.; Yin, J.; Tian, H.; Chen, S.; Wu, D.; Sun, A. TET2 mutation is unfavorable prognostic factor in cytogenetically normal acute myeloid leukemia patients with NPM1+ and FLT3-ITD- mutations. Int. J. Hematol. 2014, 100, 96–104. [Google Scholar] [CrossRef]
- Stasik, S.; Eckardt, J.N.; Kramer, M.; Rollig, C.; Kramer, A.; Scholl, S.; Hocchaus, A.; Crysandt, M.; Brummanford, T.H.; Naumann, R.; et al. Impact of PTPN11 mutations in clinical outcome analyzed in 1529 patients with acute myeloid leukemia. Blood Adv. 2011, 5, 3279–3289. [Google Scholar] [CrossRef]
- Fobare, S.; Kohlschmidt, J.; Ozer, H.G.; Mrozek, K.; Nicolet, D.; Mims, A.S.; Garzon, R.; Blachly, J.S.; Orwick, S.; Carroll, A.J.; et al. Molecular, clinical and prognostic implications of PTPN11 mutations in acute myeloid leukemia. Blood Adv. 2022, 6, 1371–1380. [Google Scholar] [CrossRef]
- Alfayez, M.; Issa, G.C.; Patel, K.P.; Wang, F.; Wang, X.; Short, N.J.; Cortes, J.E.; Kadia, T.; Ravandi, F.; Pierce, S.; et al. The clinical impact of PTPN11 mutations in adults with acute myeloid leukemia. Leukemia 2021, 35, 691–700. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, F.; Huo, L.; Cai, W.; Wang, Q.; Wen, L.; Yan, L.; Shen, H.; Xu, X.; Chen, S. Clinical characteristics and prognostic analysis of acute myeloiod leukemia patients with PTPN11 mutations. Hematology 2022, 27, 1184–1190. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Rotrhenberg-Thurley, M.; Gorlich, D.; Sauerland, M.C.; Dufour, A.M.; Schneider, S.; Subkelewe, M.; Braess, J.; Wormann, B.J.; Wormann, W.E.; et al. PTPN11 mutations and outcome in adult patients with acute myeloid leukemia. Blood 2020, 136 (Suppl. 1), 4–5. [Google Scholar] [CrossRef]
- Mrozek, K.; Marcucci, G.; Nicolet, D. Prognostic significance of the Uropean Leukemia Net standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 4515–4523. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A.; Hou, J.Z.; Fiedler, J.W.; Lin, T.L.; Walter, R.B. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: Results from a phase Ib/Ii study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Wei, A.H.; Montesinos, P.; Inanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K. Venetoclax plus Idac for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomised placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.C.; Roberts, A.W.; Reynolds, I.; Fong, C.Y.; Ting, S.B.; Salmon, J.M. Chemotherapy and venetoclax in elderly acute myeloid leukemia trial (Caveat): A phase Ib dose-escalation study of venetoclax combined with modified intensive chemotherapy. J. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loighavi, S.; Xiao, L.; Kadia, T. Venetoclax combined with Fag-Ida induction and consolidation in newly diagnosed and relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2021, 39, 2768–2778. [Google Scholar] [CrossRef]
- Chua, C.C.; Hammond, D.; Kent, A.; Tiong, I.S.; Konopleva, M.; Pollyea, D.A.; DiNardo, C.D.; Wei, A.H. Treatment-free remission after ceasing venetoclax-based therapy in patients with acute myeloid leukemia. Blood Adv. 2022, 6, 3879–3885. [Google Scholar] [CrossRef]
- Di Nardo, C.; Lachowiez, C. Acute myeloid leukemia: From mutation profiling to treatment decisions. Curr. Hematol. Malig. Rep. 2019, 14, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Bidikian, A.; Venugopal, S.; Konopleva, M.; DiNardo, C.D.; Kadia, T.M.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Yilmaz, M.; et al. Clinical outcomes associated with NPM1 mutations in patients with relapsed or refractory AML. Blood Adv. 2023, 7, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Paschka, P.; Krzyalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T. Gentuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: Early results from the prospective. Randomized AMLSG-09-09. Phase III study. J. Clin. Oncol. 2020, 38, 623–632. [Google Scholar] [CrossRef]
- Kapp-Schwoerer, S.; Weber, D.; Carbacioglu, A.; Gaidzik, V.I.; Pashka, P.; Kronke, J.; Theis, F.; Rucker, F.G.; Teleanu, M.V.; panina, E.; et al. Impact of gemtuzumab ozogamicin. On MRD and relapse risk in patients with NPM1-mutated AML: Results from the AMLSG 09-09 trial. Blood 2020, 136, 3041–3050. [Google Scholar] [CrossRef]
- Dohner, H.; Weber, D.; Krzykalla, J.; Fiedler, W.; Kuhn, M.; Schroeder, T.; Mayer, K.; Lubbert, M.; Watted, M.; Gotze, K.; et al. Gemtuzumab ozogamicin plus intensive chemotherapy for patients. With NPM1-mutated acute myeloid leukemia. Blood 2022, 140 (Suppl. 1), 218. [Google Scholar] [CrossRef]
- Dohner, H.; Weber, D.; Krzykalla, J.; Fiedler, W.; Kuhn, M.; Schroeder, T.; Mayer, K.; Lubbert, M.; Watted, M.; Gotze, K.; et al. Intensive chemotherapy with or without gemtuzumab ozogamicin in patients with NPM1-mutated acute myeloid leukemia (AMLSG 09-09): A randomised, open-lebol, munlticentre, phase 3 trial. Lancet Haematol. 2023, in press. [Google Scholar] [CrossRef]
- Russell, N.H.; Wilhelm-Benarizi, C.; Knapp, S.; Batten, L.M.; Canham, J.; Hinson, E.L.; Overgaad, U.M.; Gilkes, A.; Othman, J.; Potter, N.; et al. PLAG-Ida combined with gemtuzumab ozogamicin (GO) improves event free survival in younger patients with newly diagnosed acute myeloid leukemia (AML) and shows an overall survival benefit in NPM1 and FLT3 mutated subgroups. Results fgrom the UK NCRI AML 19 trial. Blood 2022, 140 (Suppl. 1), 526–528. [Google Scholar]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, P.; Chang, C.L.; Zhong, S.Z.; Zhu, H.H. Targetd therapy in NPM1-mutated AML: Knowns and unknowns. Front. Oncol. 2022, 12, 972606. [Google Scholar] [CrossRef] [PubMed]
- Kühn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in Npm1 mutant leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [Green Version]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y. Therapeutic targeting of preleukemia cells in a mouse model of Npmn1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.; nMcGeehan, G. Menin inhibition decreases bcl-2 and synergizes with venetoclax in Npm1/Flt3-mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schonfeld, J.; Kunz, K. Synergistic trargeting of Flt3 mutations in AML via combined menin-MLL and Flt3 inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef]
- Miao, H.; Kim, E.; Chen, D.; Purohit, T.; Kempinska, K.; Ropa, J. Combinatoria treatment with menin and Flt3 inhibitors induces complete remission in AML models with activating Flt3 mutations. Blood 2020, 136, 2958–2963. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.; Dzama, M.M.; Dolgikh, N.; Stiller, H.L.; Bohl, S.R.; Lahrmann, C.; Kunz, K.; Kessler, L.; Echchannaoui, H.; Chen, C.W.; et al. Menin inhibitor ziftomenib (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax. Haematologica 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Das, K.; Takahashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor. Ziftomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar] [CrossRef]
- Wang, E.R.S.; Altman, J.K.; Pettit, K.; De Botton, S.; Walter, R.P.; Fenaux, P. Preliminary data on a phase 1/2a first in human study of the menin-Kmt2A (MLL) inhibitor KO-539 in patients with relapsed or refractory acute myeloid leukemia. Blood 2020, 136 (Suppl. 1), 7–8. [Google Scholar] [CrossRef]
- Stein, E.M.; Aldoss, I.; DiPersio, J.F.; Stone, R.M.; Arellano, M.L.; Rosen, G. Safety and efficacy of menin inhibition in patients (Pts) with MLL-rearranged and NPM1 mutant acute acute leukemia: A phase (Ph) 1, first-in-human study of SNDX-5613 (Augment 101). Blood 2021, 138 (Suppl. 1), 699. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cugklievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Pater, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukemia. Nature 2023, 615, 920–925. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef]
- Erba, H.P.; Fathi, A.T.; Issa, G.C.; Altman, J.K.; Montesinos, P.; Patnaik, M.M.; Foran, J.M.; De Botton, S.; Baer, M.R.; Schiller, G.J.; et al. Update of a phase 1–2 first-in-human study of the menin-KMT2A (MLL) inhibitor ziftomenib (KO-539) in patients with relapsed or refractory acute myeloid leukemia. Blood 2022, 140 (Suppl. 1), 153–156. [Google Scholar] [CrossRef]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.; Garcia-Manewo, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-mutated myelodysplastic syndrome and acute myeloid leukemia: Biology, current therapy, and future directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.; Douglas, L.R.; Duriez, P.J.; Balourdas, D.I.; Joerger, A.C.; Khadiullina, R.; Bulatov, E.; Baud, M. Discovery of nanomolar-affinity pharmacological chaperones stabilizing the oncogenic p53 mutant Y220C. ACS Pharmacol. Transl. Sci. 2022, 5, 1169–1180. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Pelosi, E.; Castelli, G. Genetic, Phenotypic, and Clinical Heterogeneity of NPM1-Mutant Acute Myeloid Leukemias. Biomedicines 2023, 11, 1805. https://doi.org/10.3390/biomedicines11071805
Testa U, Pelosi E, Castelli G. Genetic, Phenotypic, and Clinical Heterogeneity of NPM1-Mutant Acute Myeloid Leukemias. Biomedicines. 2023; 11(7):1805. https://doi.org/10.3390/biomedicines11071805
Chicago/Turabian StyleTesta, Ugo, Elvira Pelosi, and Germana Castelli. 2023. "Genetic, Phenotypic, and Clinical Heterogeneity of NPM1-Mutant Acute Myeloid Leukemias" Biomedicines 11, no. 7: 1805. https://doi.org/10.3390/biomedicines11071805
APA StyleTesta, U., Pelosi, E., & Castelli, G. (2023). Genetic, Phenotypic, and Clinical Heterogeneity of NPM1-Mutant Acute Myeloid Leukemias. Biomedicines, 11(7), 1805. https://doi.org/10.3390/biomedicines11071805