

Aldosterone Contributes to Vasopressin Escape through Changes in Water and Urea Transport

 and
and 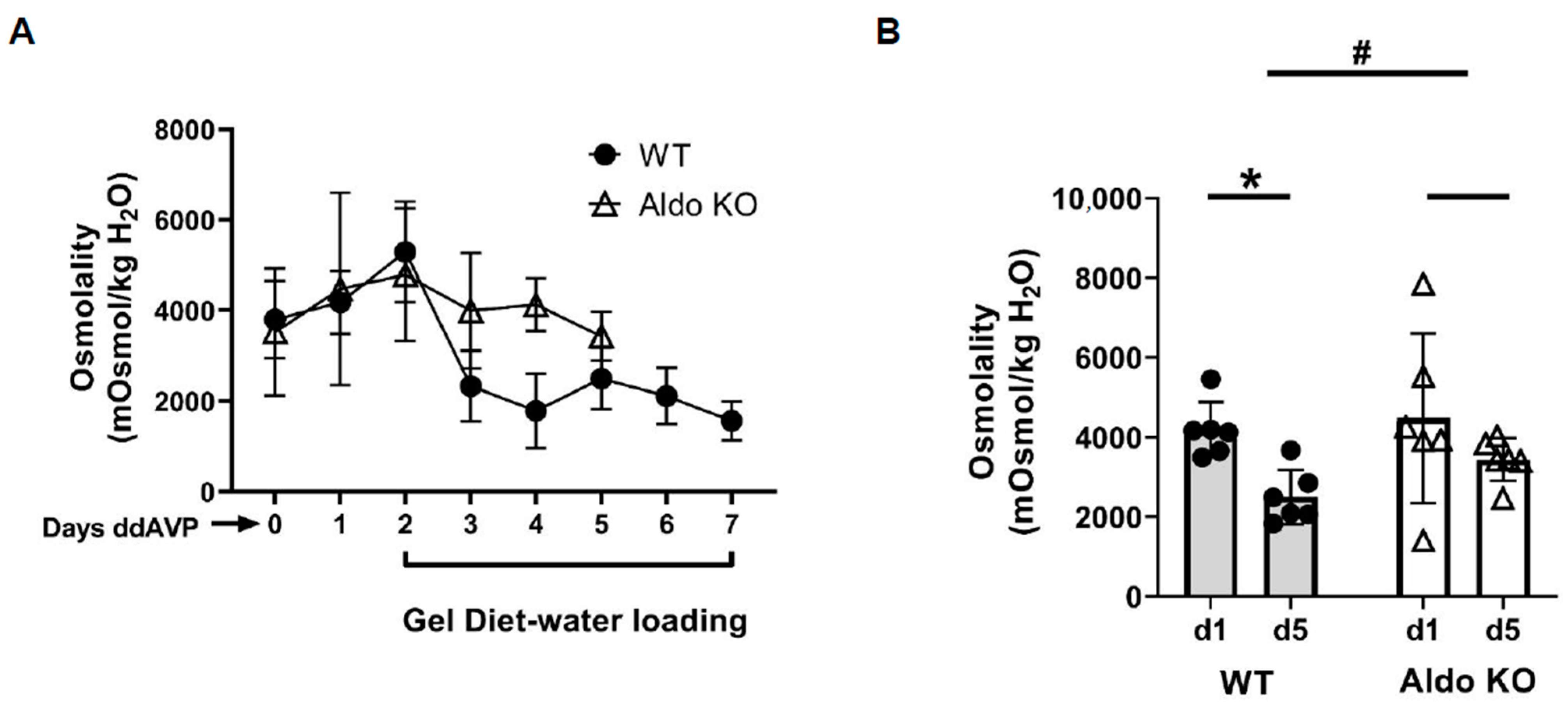{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Vasopressin Escape Protocol
2.3. Western Blot Analysis
2.4. Statistics
3. Results
3.1. Vasopressin Escape Was Observed in Wild-Type but Not in Aldosterone Synthase Knockout Mice
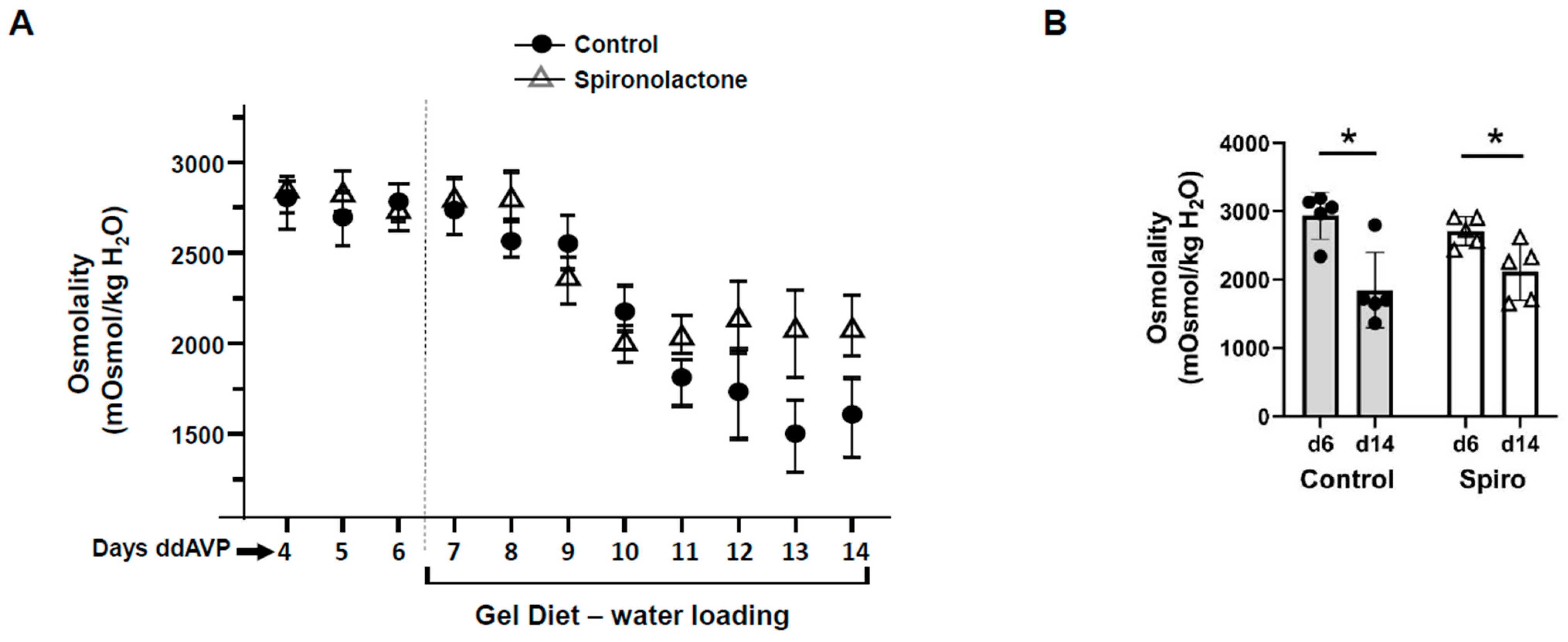
3.2. Inhibition of the Mineralocorticoid Receptor (MR) Blunted Vasopressin Escape
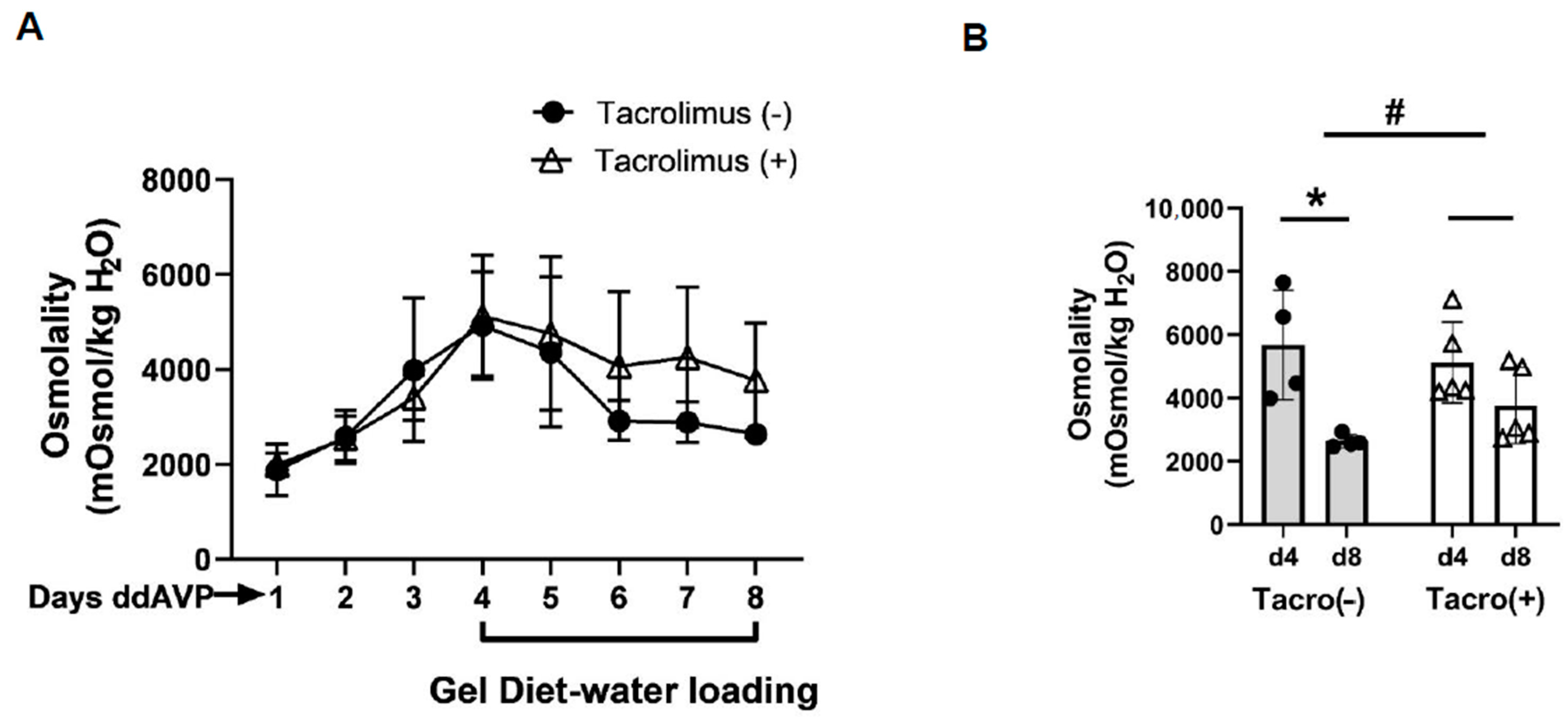
3.3. Inhibition of Calcineurin Diminished Vasopressin Escape
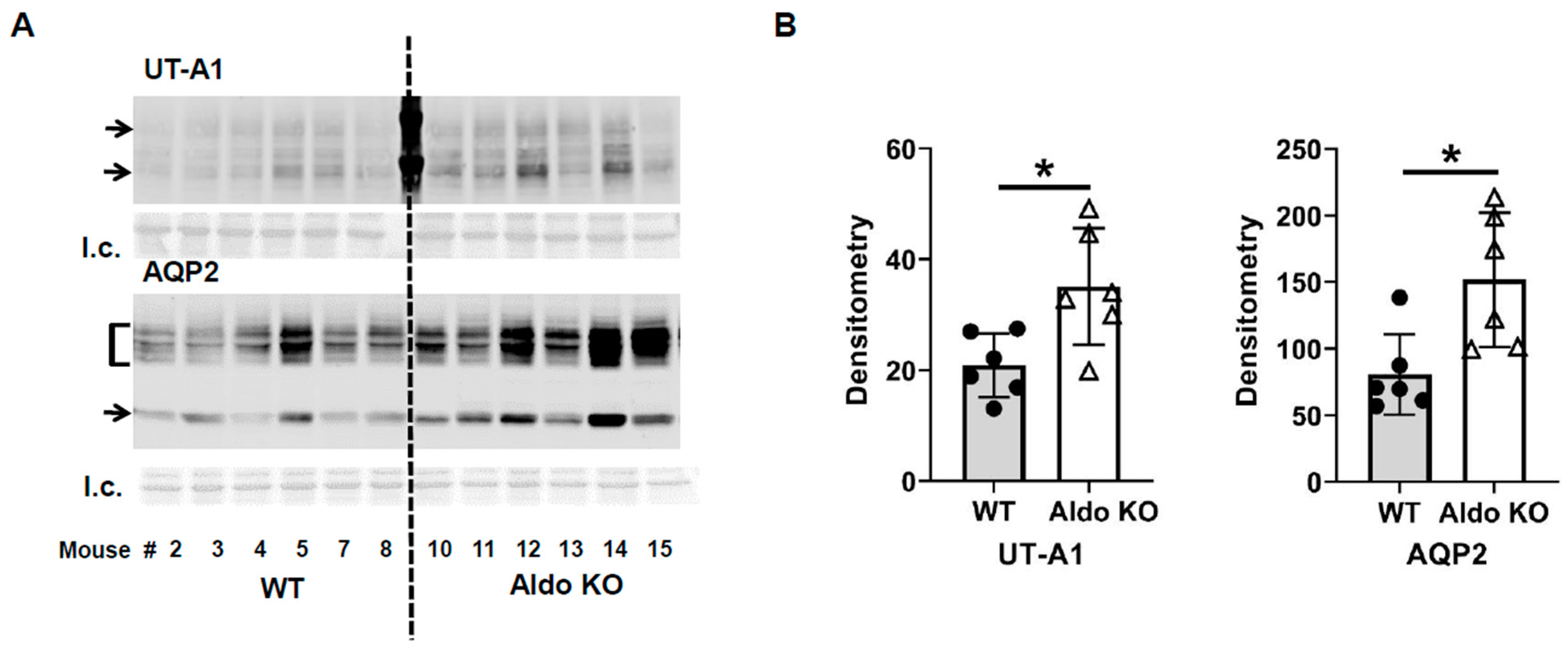
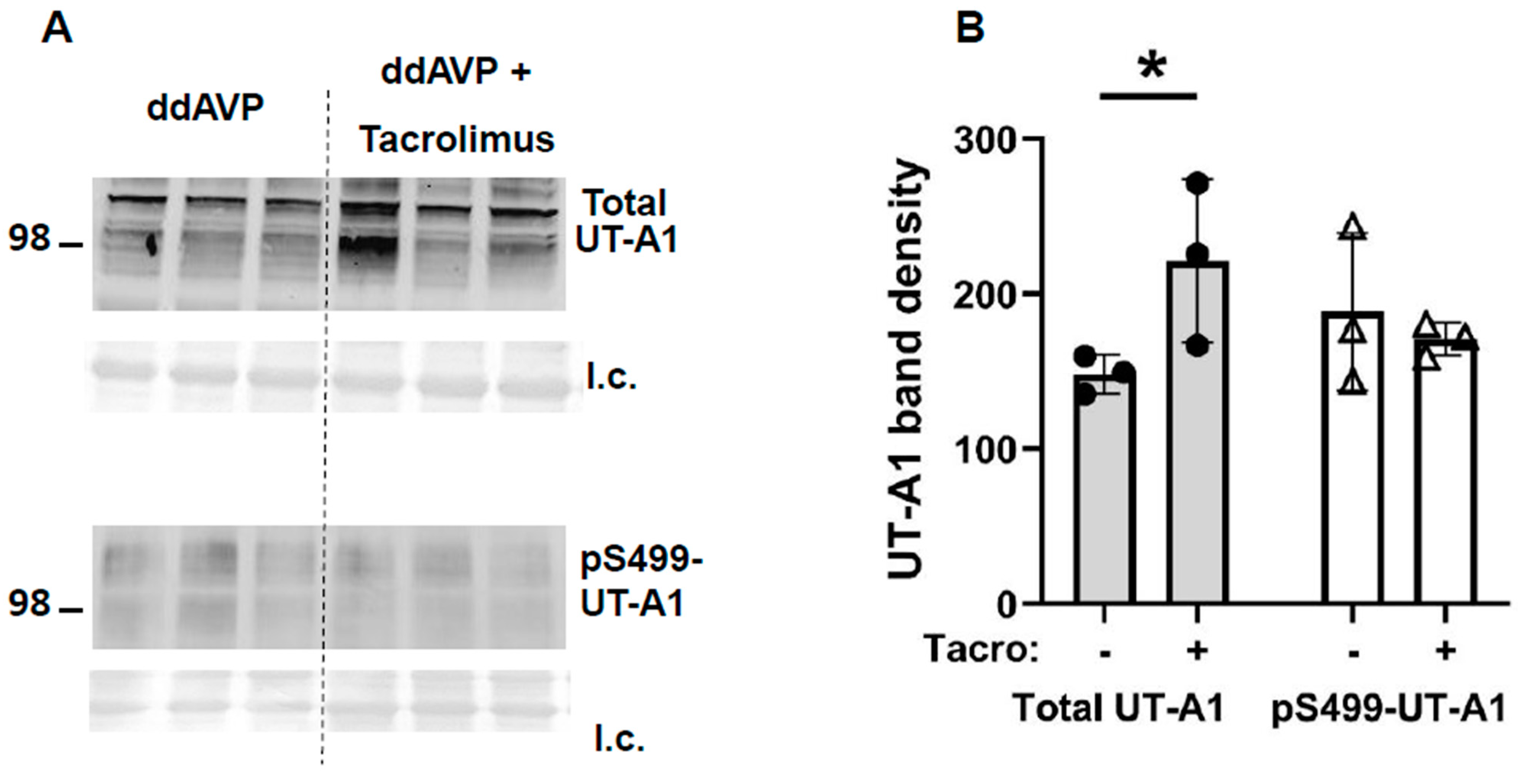
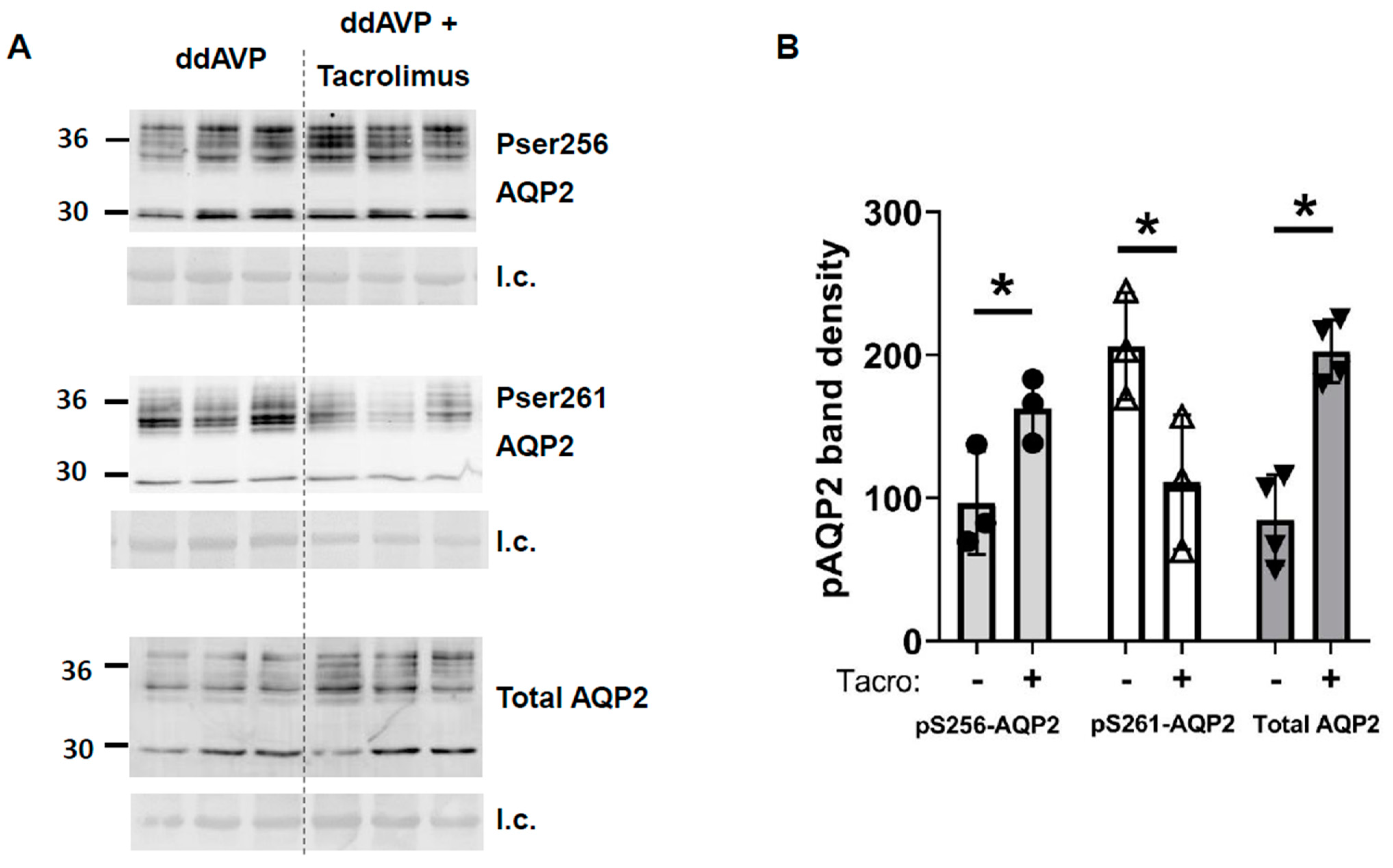
3.4. Inhibition of Calcineurin Increased UT-A1 and AQP2 Protein Abundances during Vasopressin Escape
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berl, T.S.J. Disorders of water metabolism. In Comprehensive Clinical Nephrology, 6th ed.; Elsevier: Philadelphia, PA, USA, 2019; pp. 94–110. [Google Scholar]
- Verbalis, J.G.; Berl, T. Disorders of water balance. In The Kidney; Brenner, B.M., Ed.; Saunders: Philadelphia, PA, USA, 2008; pp. 459–549. [Google Scholar]
- Ecelbarger, C.A.; Murase, T.; Tian, Y.; Nielsen, S.; Knepper, M.A.; Verbalis, J.G. Regulation of renal salt and water transporters during vasopressin escape. Prog. Brain Res. 2002, 139, 75–84. [Google Scholar]
- Chou, C.L.; Yip, K.P.; Michea, L.; Kador, K.; Ferraris, J.D.; Wade, J.B.; Knepper, M.A. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct—Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 2000, 275, 36839–36846. [Google Scholar] [CrossRef] [Green Version]
- Knepper, M.A.; Inoue, T. Regulation of aquaporin-2 water channel trafficking by vasopressin. Curr. Opin. Cell Biol. 1997, 9, 560–564. [Google Scholar] [CrossRef]
- Sands, J.M.; Nonoguchi, H.; Knepper, M.A. Vasopressin effects on urea and H2O transport in inner medullary collecting duct subsegments. Am. J. Physiol. 1987, 253, F823–F832. [Google Scholar] [CrossRef] [PubMed]
- Himmel, N.J.; Rogers, R.T.; Redd, S.K.; Wang, Y.; Blount, M.A. Purinergic signaling is enhanced in the absence of UT-A1 and UT-A3. Physiol. Rep. 2021, 9, e14636. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Hasler, U.; Nunes, P.; Bouley, R.; Lu, H. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr. Opin. Nephrol. Hypertens. 2010, 17, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Hoffert, J.D.; Nielsen, J.; Yu, M.J.; Pisitkun, T.; Schleicher, S.M.; Nielsen, S.; Knepper, M.A. Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am. J. Physiol. Renal Physiol. 2007, 292, F691–F700. [Google Scholar] [CrossRef]
- Blount, M.A.; Mistry, A.C.; Fröhlich, O.; Price, S.R.; Chen, G.; Sands, J.M.; Klein, J.D. Phosphorylation of UT-A1 urea transporter at serines 486 and 499 is important for vasopressin-regulated activity and membrane accumulation. Am. J. Physiol. Renal Physiol. 2008, 295, F295–F299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecelbarger, C.A.; Nielsen, S.; Olson, B.R.; Murase, T.; Baker, E.A.; Knepper, M.A.; Verbalis, J.G. Role of renal aquaporins in escape from vasopressin-induced antidiuresis in rat. J. Clin. Investig. 1997, 99, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Klein, J.D.; Sands, J.M. Phosphatases Decrease Water and Urea Permeability in Rat Inner Medullary Collecting Ducts. Int. J. Mol. Sci. 2023, 24, 6537. [Google Scholar] [CrossRef]
- Song, J.; Hu, X.Q.; Khan, O.; Tian, Y.; Verbalis, J.G.; Ecelbarger, C.A. Increased blood pressure, aldosterone activity, and regional differences in renal ENaC protein during vasopressin escape. Am. J. Physiol. Renal Physiol. 2004, 287, F1076–F1083. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Packer, R.K.; Hu, X.Q.; Sugimura, Y.; Verbalis, J.G.; Ecelbarger, C.A. Increased renal alpha-ENaC and NCC abundance and elevated blood pressure are independent of hyperaldosteronism in vasopressin escape. Am. J. Physiol. Renal Physiol. 2006, 291, F49–F57. [Google Scholar] [CrossRef] [Green Version]
- Terker, A.S.; Ellison, D.H. Renal mineralocorticoid receptor and electrolyte homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R1068–R1070. [Google Scholar] [CrossRef] [Green Version]
- Fuller, P.J.; Young, M.J. Mechanisms of mineralocorticoid action. Hypertension 2005, 46, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.R.; Stoner, L.C.; Viggiano, S.C.; Angelides, K.J.; Benos, D.J. Effects of vasopressin and aldosterone on the lateral mobility of epithelial Na+ channels in A6 renal epithelial cells. J. Membr. Biol. 1995, 147, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.H.; Menouar, M.A.; Dunn, R.J. Physiology, Aldosterone. In StatPearls. Treasure Island (FL). Available online: https://www.ncbi.nlm.nih.gov/books/NBK470339/ (accessed on 23 June 2023).
- Tumlin, J.A.; Lea, J.P.; Swanson, C.E.; Smith, C.L.; Edge, S.S.; Someren, J.S. Aldosterone and dexamethasone stimulate calcineurin activity through a transcription-independent mechanism involving steroid receptor-associated heat shock proteins. J. Clin. Investig. 1997, 99, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Good, D.W. Nongenomic actions of aldosterone on the renal tubule. Hypertension 2007, 49, 728–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Yang, B.; Ruiz, J.A.; Efe, O.; Ilori, T.O.; Sands, J.M.; Klein, J.D. Phosphatase inhibition increases AQP2 accumulation in the rat IMCD apical plasma membrane. Am. J. Physiol. Renal Physiol. 2016, 311, F1189–F1197. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Alsady, M.; Chou, C.L.; de Groot, T.; Deen, P.M.T.; Knepper, M.A.; Ecelbarger, C.M. Single-tubule RNA-Seq uncovers signaling mechanisms that defend against hyponatremia in SIADH. Kidney Int 2018, 93, 128–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Makhanova, N.; Caron, K.; Lopez, M.L.; Gomez, R.A.; Smithies, O.; Kim, H.S. Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 2005, 146, 2650–2656. [Google Scholar] [CrossRef] [PubMed]
- Knepper, M.A.; Good, D.W.; Burg, M.B. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am. J. Physiol. 1985, 249, F870–F877. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.L.; Hwang, G.; Hageman, D.J.; Han, L.; Agrawal, P.; Pisitkun, T.; Knepper, M.A. Identification of UT-A1- and AQP2-interacting proteins in rat inner medullary collecting duct. Am. J. Physiol. Cell. Physiol. 2018, 314, C99–C117. [Google Scholar] [CrossRef] [PubMed]
- De Seigneux, S.; Nielsen, J.; Olesen, E.T.; Dimke, H.; Kwon, T.H.; Frokiaer, J.; Nielsen, S. Long-term aldosterone treatment induces decreased apical but increased basolateral expression of AQP2 in CCD of rat kidney. Am. J. Physiol. Renal Physiol. 2007, 293, F87–F99. [Google Scholar] [CrossRef]
- Lee, B.H.; Kwon, T.H. Regulation of AQP2 in Collecting Duct: An emphasis on the Effects of Angiotensin II or Aldosterone. Electrolytes Blood Press. 2007, 5, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Poulsen, S.B.; Wu, Q.; Esteva-Font, C.; Olesen, E.T.B.; Peng, L.; Olde, B.; Leeb-Lundberg, L.M.F.; Pisitkun, T.; Rieg, T.; et al. Rapid Aldosterone-Mediated Signaling in the DCT Increases Activity of the Thiazide-Sensitive NaCl Cotransporter. J. Am. Soc. Nephrol. 2019, 30, 1454–1470. [Google Scholar] [CrossRef] [PubMed]
- Pergher, P.S.; Leite-Dellova, D.; de Mello-Aires, M. Direct action of aldosterone on bicarbonate reabsorption in in vivo cortical proximal tubule. Am. J. Physiol. Renal Physiol. 2009, 296, F1185–F1193. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; LaRocque, L.M.; Ruiz, J.A.; Rodriguez, E.L.; Sands, J.M.; Klein, J.D. Aldosterone Contributes to Vasopressin Escape through Changes in Water and Urea Transport. Biomedicines 2023, 11, 1844. https://doi.org/10.3390/biomedicines11071844
Wang Y, LaRocque LM, Ruiz JA, Rodriguez EL, Sands JM, Klein JD. Aldosterone Contributes to Vasopressin Escape through Changes in Water and Urea Transport. Biomedicines. 2023; 11(7):1844. https://doi.org/10.3390/biomedicines11071844
Chicago/Turabian StyleWang, Yanhua, Lauren M. LaRocque, Joseph A. Ruiz, Eva L. Rodriguez, Jeff M. Sands, and Janet D. Klein. 2023. "Aldosterone Contributes to Vasopressin Escape through Changes in Water and Urea Transport" Biomedicines 11, no. 7: 1844. https://doi.org/10.3390/biomedicines11071844
APA StyleWang, Y., LaRocque, L. M., Ruiz, J. A., Rodriguez, E. L., Sands, J. M., & Klein, J. D. (2023). Aldosterone Contributes to Vasopressin Escape through Changes in Water and Urea Transport. Biomedicines, 11(7), 1844. https://doi.org/10.3390/biomedicines11071844