
Interaction of Brain-Derived Neurotrophic Factor with the Effects of Chronic Methamphetamine on Prepulse Inhibition in Mice Is Independent of Dopamine D3 Receptors
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
- Step 1:
- Crossing of BDNF HET with D3 knockout mice, generating 50% BDNF HET/D3 HET and 50% BDNF wildtype/D3 HET mice with identical genetic background. These animals were used as founding breeders of the double-mutant line.
- Step 2:
- Mating BDNF HET/D3 receptor HET mice with BDNF wildtype/D3 receptor HET, generating eight possible genetic combinations of which four were used in the present study (Supplementary Figure S1): WT, BDNF HET, D3KO and DM. The numbers of mice per group are shown in Table 1. For both breeding strategy steps, the genotype of male and female breeders was randomised to account for possible differences in maternal behaviour.
2.2. Chronic METH Treatment
2.3. Behavioural Analysis
2.3.1. Prepulse Inhibition
2.3.2. Locomotor Hyperactivity
2.4. Quantitative Reverse Transcriptase-PCR
- beta actin: F: 5′-GATCATTGCTCCTCCTGAGC-3′, R: 5′-AGTCCGCCTAGAAGCACTTG-3′
- D1 receptor: F: 5′-CCAGATCGGGCATTTGGAGA-3′, R: 5′-GGGCCTCTTCCTGGTCAATC-3′
- D2 receptor: F: 5′-GTCTCGTTCTACGTGCCCTT-3′, R: 5′-GGTGGGTACAGTTGCCCTTG-3′
- D3 receptor: F: 5′-ACTTGGAGGTGACAGGTGGA-3′, R: 5′-GGCATGACCACTGCTGTGTA-3′
- DAT: F: 5′-CCTGGTTCTACGGTGTCCAG-3′, R: 5′-GCTGACCACGACCACATACA-3′.
2.5. Data Analysis
3. Results
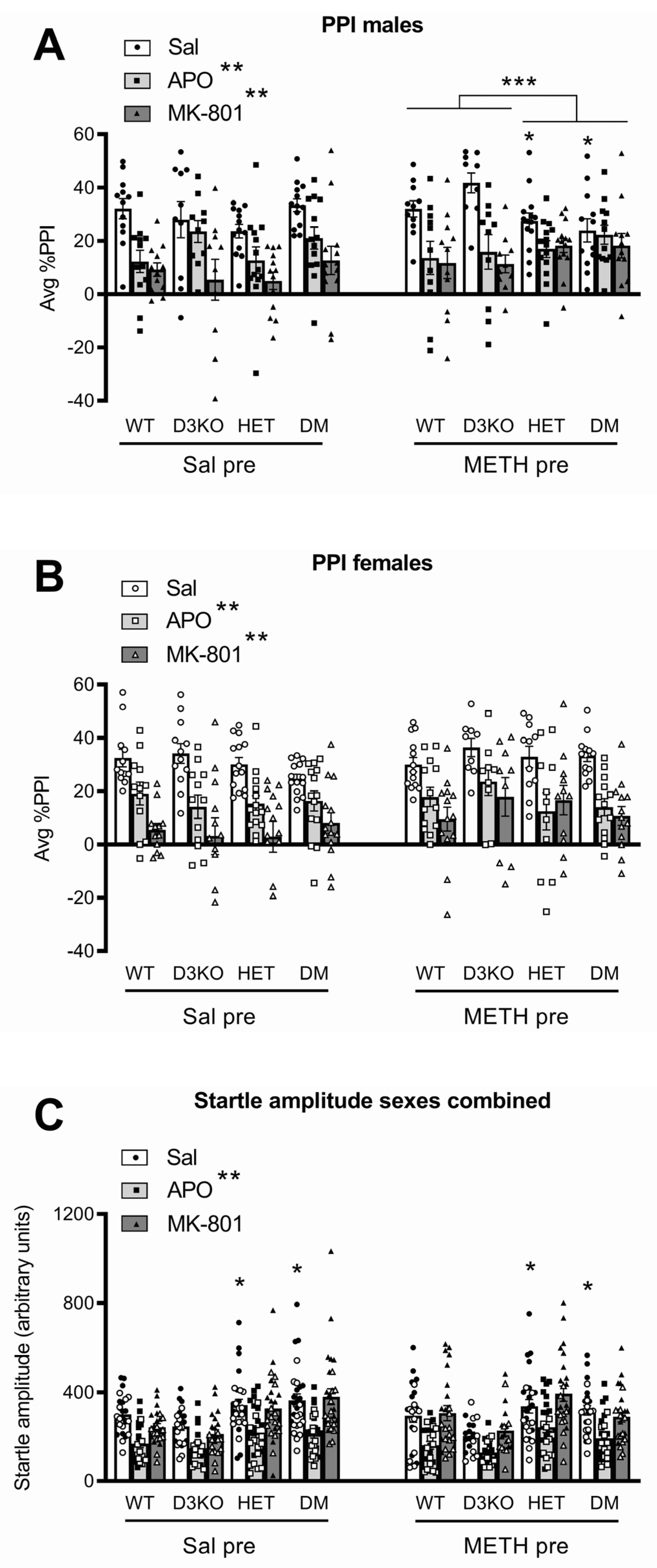
3.1. Baseline PPI and Startle Are Affected by BDNF Genotype but Not by D3 Receptor Deletion
3.2. BDNF Haploinsufficiency, but Not D3 Receptor Knockout, Reduces APO-Induced PPI Disruption in METH-Sensitised Male Mice
3.3. BDNF Haploinsufficiency, but Not D3 Receptor Knockout, Reduces MK-801-Induced PPI Disruption in METH-Sensitised Male Mice
3.4. Female BDNF HET Mice Show Endogenous Sensitisation, and Chronic METH Induces Further Long-Term Locomotor Sensitisation Independent of BDNF or D3 Receptor Genotype
3.5. Sex-Specific Downregulation of Dopamine Receptor Gene Expression in BDNF HET Mice in the Striatum Independent of METH Pretreatment
3.6. Differential Effects of Chronic METH on DAT Expression in Striatum Dependent on Sex, D3 Receptor and BDNF Genotype
3.7. Sex-Specific Downregulation of Dopamine Receptor Expression in BDNF HET Mice in the Frontal Cortex
3.8. Differential Effects of Chronic METH on DAT Expression in Frontal Cortex Dependent on Sex and D3 Receptor Genotype
4. Discussion
4.1. Chronic METH Affects Baseline PPI and Drug-Induced PPI Disruption in Male BDNF HET Mice Independent of D3 Receptors
4.2. Chronic METH Induces Locomotor Hyperactivity Sensitisation Independent of D3 Receptors
4.3. Sex-Specific Changes in Dopamine Receptor and Dopamine Transporter Gene Expression in BDNF HET Mice in the Striatum and Frontal Cortex
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hogarth, S.; Manning, E.E.; van den Buuse, M. Chronic methamphetamine and psychosis pathways. In Handbook of Substance Misuse and Addictions; Patel, V.B., Preedy, V.R., Eds.; Springer: Cham, Switzerland, 2022; pp. 2121–2146. [Google Scholar] [CrossRef]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; Garcia-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef] [PubMed]
- McKetin, R.; Hickey, K.; Devlin, K.; Lawrence, K. The risk of psychotic symptoms associated with recreational methamphetamine use. Drug Alcohol. Rev. 2010, 29, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.C.; Kalivas, P.W. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res. Brain Res. Rev. 1997, 25, 192–216. [Google Scholar] [CrossRef]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M. The role of endogenous sensitization in the pathophysiology of schizophrenia: Implications from recent brain imaging studies. Brain Res. Brain Res. Rev. 2000, 31, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Weidenauer, A.; Bauer, M.; Sauerzopf, U.; Bartova, L.; Nics, L.; Pfaff, S.; Philippe, C.; Berroteran-Infante, N.; Pichler, V.; Meyer, B.M.; et al. On the relationship of first-episode psychosis to the amphetamine-sensitized state: A dopamine D2/3 receptor agonist radioligand study. Transl. Psychiatry 2020, 10, 2. [Google Scholar] [CrossRef]
- Weidenauer, A.; Bauer, M.; Sauerzopf, U.; Bartova, L.; Praschak-Rieder, N.; Sitte, H.H.; Kasper, S.; Willeit, M. Making Sense of: Sensitization in Schizophrenia. Int. J. Neuropsychopharmacol. 2017, 20, 1–10. [Google Scholar] [CrossRef]
- Notaras, M.; van den Buuse, M. Brain-Derived Neurotrophic Factor (BDNF): Novel Insights into Regulation and Genetic Variation. Neuroscientist 2019, 25, 434–454. [Google Scholar] [CrossRef]
- Tsai, S.J. Critical issues in BDNF Val66Met genetic studies of neuropsychiatric disorders. Front. Mol. Neurosci. 2018, 11, 156. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef]
- Ikemoto, S. Brain reward circuitry beyond the mesolimbic dopamine system: A neurobiological theory. Neurosci. Biobehav. Rev. 2010, 35, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wolf, M.E. Multiple faces of BDNF in cocaine addiction. Behav. Brain Res. 2015, 279, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Logrip, M.L.; Barak, S.; Warnault, V.; Ron, D. Corticostriatal BDNF and alcohol addiction. Brain Res. 2015, 1628, 60–67. [Google Scholar] [CrossRef]
- Xu, X.; Ji, H.; Liu, G.; Wang, Q.; Liu, H.; Shen, W.; Li, L.; Xie, X.; Zhou, W.; Duan, S. A significant association between BDNF promoter methylation and the risk of drug addiction. Gene 2016, 584, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef]
- Manning, E.E.; Halberstadt, A.L.; van den Buuse, M. BDNF-deficient mice show reduced psychosis-related behaviors following chronic methamphetamine. Int. J. Neuropsychopharmacol. 2016, 19, pyv116. [Google Scholar] [CrossRef]
- Notaras, M.; Hill, R.; van den Buuse, M. A role for the BDNF gene Val66Met polymorphism in schizophrenia? A comprehensive review. Neurosci. Biobehav. Rev. 2015, 51, 15–30. [Google Scholar] [CrossRef]
- Notaras, M.; Hill, R.; van den Buuse, M. The BDNF gene Val66Met polymorphism as a modifier of psychiatric disorder susceptibility: Progress and controversy. Mol. Psychiatry 2015, 20, 916–930. [Google Scholar] [CrossRef]
- Gururajan, A.; Hill, R.A.; van den Buuse, M. Brain-derived neurotrophic factor heterozygous mutant rats show selective cognitive changes and vulnerability to chronic corticosterone treatment. Neuroscience 2015, 284, 297–310. [Google Scholar] [CrossRef]
- Hill, R.A.; van den Buuse, M. Sex-dependent and region-specific changes in TrkB signaling in BDNF heterozygous mice. Brain Res. 2011, 1384, 51–60. [Google Scholar] [CrossRef]
- Graham, D.L.; Edwards, S.; Bachtell, R.K.; DiLeone, R.J.; Rios, M.; Self, D.W. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat. Neurosci. 2007, 10, 1029–1037. [Google Scholar] [CrossRef]
- Birbeck, J.A.; Khalid, M.; Mathews, T.A. Potentiated striatal dopamine release leads to hyperdopaminergia in female brain-derived neurotrophic factor heterozygous mice. ACS Chem. Neurosci. 2014, 5, 275–281. [Google Scholar] [CrossRef]
- Saylor, A.J.; McGinty, J.F. Amphetamine-induced locomotion and gene expression are altered in BDNF heterozygous mice. Genes. Brain Behav. 2008, 7, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Richtand, N.M.; Woods, S.C.; Berger, S.P.; Strakowski, S.M. D3 dopamine receptor, behavioral sensitization, and psychosis. Neurosci. Biobehav. Rev. 2001, 25, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Wang, Y.M.; Miles, P.R.; Budygin, E.A.; Picetti, R.; Gainetdinov, R.R.; Caron, M.G.; Wightman, R.M. Dopamine autoreceptor regulation of release and uptake in mouse brain slices in the absence of D(3) receptors. Neuroscience 2002, 112, 39–49. [Google Scholar] [CrossRef]
- Le Foll, B.; Diaz, J.; Sokoloff, P. Neuroadaptations to hyperdopaminergia in dopamine D3 receptor-deficient mice. Life Sci. 2005, 76, 1281–1296. [Google Scholar] [CrossRef]
- Zapata, A.; Kivell, B.; Han, Y.; Javitch, J.A.; Bolan, E.A.; Kuraguntla, D.; Jaligam, V.; Oz, M.; Jayanthi, L.D.; Samuvel, D.J.; et al. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors. J. Biol. Chem. 2007, 282, 35842–35854. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kuzhikandathil, E.V. Molecular characterization of individual D3 dopamine receptor-expressing cells isolated from multiple brain regions of a novel mouse model. Brain Struct. Funct. 2012, 217, 809–833. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.; Pfeifer, S.; Schaub, R.T.; Grabe, H.J.; Barnow, S.; Freyberger, H.J.; Cascorbi, I. Decreased levels of dopamine D3 receptor mRNA in schizophrenic and bipolar patients. Neuropsychobiology 2004, 50, 305–310. [Google Scholar] [CrossRef]
- Schmauss, C.; Haroutunian, V.; Davis, K.L.; Davidson, M. Selective loss of dopamine D3-type receptor mRNA expression in parietal and motor cortices of patients with chronic schizophrenia. Proc. Natl. Acad. Sci. USA 1993, 90, 8942–8946. [Google Scholar] [CrossRef]
- Schmauss, C. Enhanced cleavage of an atypical intron of dopamine D3-receptor pre-mRNA in chronic schizophrenia. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 7902–7909. [Google Scholar] [CrossRef]
- Shaikh, S.; Collier, D.A.; Sham, P.C.; Ball, D.; Aitchison, K.; Vallada, H.; Smith, I.; Gill, M.; Kerwin, R.W. Allelic association between a Ser-9-Gly polymorphism in the dopamine D3 receptor gene and schizophrenia. Hum. Genet. 1996, 97, 714–719. [Google Scholar] [CrossRef]
- Williams, J.; Spurlock, G.; Holmans, P.; Mant, R.; Murphy, K.; Jones, L.; Cardno, A.; Asherson, P.; Blackwood, D.; Muir, W.; et al. A meta-analysis and transmission disequilibrium study of association between the dopamine D3 receptor gene and schizophrenia. Mol. Psychiatry 1998, 3, 141–149. [Google Scholar] [CrossRef]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef]
- Manning, E.E.; van den Buuse, M. BDNF deficiency and young-adult methamphetamine induce sex-specific effects on prepulse inhibition regulation. Front. Cell Neurosci. 2013, 7, 92. [Google Scholar] [CrossRef]
- Gogos, A.; van den Buuse, M. Sex differences in psychosis: Focus on animal models. Curr. Top. Behav. Neurosci. 2023, 62, 133–163. [Google Scholar] [CrossRef]
- van den Buuse, M. Modeling the positive symptoms of schizophrenia in genetically modified mice: Pharmacology and methodology aspects. Schizophr. Bull. 2010, 36, 246–270. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.B.; Zhou, X.; Geyer, M.A. Prepulse inhibition and genetic mouse models of schizophrenia. Behav. Brain Res. 2009, 204, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Weber, M.; Qu, Y.; Light, G.A.; Braff, D.L. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology 2008, 199, 331–388. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.A.; Krebs-Thomson, K.; Braff, D.L.; Swerdlow, N.R. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: A decade in review. Psychopharmacology 2001, 156, 117–154. [Google Scholar] [CrossRef] [PubMed]
- Mansbach, R.S.; Geyer, M.A.; Braff, D.L. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology 1988, 94, 507–514. [Google Scholar] [CrossRef] [PubMed]
- van den Buuse, M.; Biel, D.; Radscheit, K. Does genetic BDNF deficiency in rats interact with neurotransmitter control of prepulse inhibition? Implications for schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 192–198. [Google Scholar] [CrossRef]
- Ralph, R.J.; Varty, G.B.; Kelly, M.A.; Wang, Y.M.; Caron, M.G.; Rubinstein, M.; Grandy, D.K.; Low, M.J.; Geyer, M.A. The dopamine D2, but not D3 or D4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. J. Neurosci. 1999, 19, 4627–4633. [Google Scholar] [CrossRef]
- Doherty, J.M.; Masten, V.L.; Powell, S.B.; Ralph, R.J.; Klamer, D.; Low, M.J.; Geyer, M.A. Contributions of dopamine D1, D2, and D3 receptor subtypes to the disruptive effects of cocaine on prepulse inhibition in mice. Neuropsychopharmacology 2008, 33, 2648–2656. [Google Scholar] [CrossRef]
- Ernfors, P.; Lee, K.; Jaenisch, R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef]
- Erickson, J.T.; Conover, J.C.; Borday, V.; Champagnat, J.; Barbacid, M.; Yancopoulos, G.; Katz, D.M. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J. Neurosci. 1996, 16, 5361–5371. [Google Scholar] [CrossRef]
- McNamara, F.N.; Clifford, J.J.; Tighe, O.; Kinsella, A.; Drago, J.; Fuchs, S.; Croke, D.T.; Waddington, J.L. Phenotypic, ethologically based resolution of spontaneous and D2-like vs D1-like agonist-induced behavioural topography in mice with congenic D3 dopamine receptor “knockout”. Synapse 2002, 46, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Fishburn, C.S.; Drago, J.; Steiner, H.; Lachowicz, J.E.; Park, B.H.; Gauda, E.B.; Lee, E.J.; Cool, M.H.; Sibley, D.R.; et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 1945–1949. [Google Scholar] [CrossRef]
- Greening, D.W.; Notaras, M.; Chen, M.; Xu, R.; Smith, J.D.; Cheng, L.; Simpson, R.J.; Hill, A.F.; van den Buuse, M. Chronic methamphetamine interacts with BDNF Val66Met to remodel psychosis pathways in the mesocorticolimbic proteome. Mol. Psychiatry 2021, 26, 4431–4447. [Google Scholar] [CrossRef]
- Hume, C.; Massey, S.; van den Buuse, M. The effect of chronic methamphetamine treatment on schizophrenia endophenotypes in heterozygous reelin mice: Implications for schizophrenia. Biomolecules 2020, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Jaehne, E.J.; Ameti, D.; Paiva, T.; van den Buuse, M. Investigating the role of serotonin in methamphetamine psychosis: Unaltered behavioral effects of chronic methamphetamine in 5-HT1A knockout mice. Front. Psychiatry 2017, 8, 61. [Google Scholar] [CrossRef]
- van Nimwegen, L.; de Haan, L.; van Beveren, N.; van den Brink, W.; Linszen, D. Adolescence, schizophrenia and drug abuse: A window of vulnerability. Acta Psychiatr. Scand. 2005, 111, 35–42. [Google Scholar] [CrossRef]
- Bramness, J.G.; Gundersen, Ø.H.; Guterstam, J.; Rognli, E.B.; Konstenius, M.; Løberg, E.M.; Medhus, S.; Tanum, L.; Franck, J. Amphetamine-induced psychosis—A separate diagnostic entity or primary psychosis triggered in the vulnerable? BMC Psychiatry 2012, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.N.; Renish, L.; Osredkar, T.; Walro, J.M.; Kucera, J.; Dluzen, D.E. Methamphetamine-induced loss of striatal dopamine innervation in BDNF heterozygote mice does not further reduce D3 receptor concentrations. Synapse 2004, 52, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sbisa, A.; Kusljic, S.; Zethoven, D.; van den Buuse, M.; Gogos, A. The effect of 17beta-estradiol on maternal immune activation-induced changes in prepulse inhibition and dopamine receptor and transporter binding in female rats. Schizophr. Res. 2020, 223, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Klug, M.; Hill, R.A.; Choy, K.H.; Kyrios, M.; Hannan, A.J.; van den Buuse, M. Long-term behavioral and NMDA receptor effects of young-adult corticosterone treatment in BDNF heterozygous mice. Neurobiol. Dis. 2012, 46, 722–731. [Google Scholar] [CrossRef]
- Advani, T.; Koek, W.; Hensler, J.G. Gender differences in the enhanced vulnerability of BDNF+/− mice to mild stress. Int. J. Neuropsychopharmacol. 2009, 12, 583–588. [Google Scholar] [CrossRef]
- Du, X.; Serena, K.; Hwang, W.J.; Grech, A.M.; Wu, Y.W.C.; Schroeder, A.; Hill, R.A. Prefrontal cortical parvalbumin and somatostatin expression and cell density increase during adolescence and are modified by BDNF and sex. Mol. Cell Neurosci. 2018, 88, 177–188. [Google Scholar] [CrossRef]
- Johansen, A.; McFadden, L.M. The neurochemical consequences of methamphetamine self-administration in male and female rats. Drug Alcohol. Depend. 2017, 178, 70–74. [Google Scholar] [CrossRef]
- Chan, C.B.; Ye, K. Sex differences in brain-derived neurotrophic factor signaling and functions. J. Neurosci. Res. 2017, 95, 328–335. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Larson, G.A.; Swant, J.; Sen, N.; Javitch, J.A.; Zahniser, N.R.; De Felice, L.J.; Khoshbouei, H. Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J. Biol. Chem. 2009, 284, 2978–2989. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Koeltzow, T.E.; Santiago, G.T.; Moratalla, R.; Cooper, D.C.; Hu, X.T.; White, N.M.; Graybiel, A.M.; White, F.J.; Tonegawa, S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron 1997, 19, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Griffon, N.; Diaz, J.; Le Foll, B.; Bezard, E.; Gross, C.; Lammers, C.; Stark, H.; Carroll, P.; Schwartz, J.C.; et al. Brain-derived neurotrophic factor and the plasticity of the mesolimbic dopamine pathway. Int. Rev. Neurobiol. 2004, 59, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, C.; Wiedermann, D.; Neumaier, B.; Drzezga, A.; Timmermann, L.; Graf, R.; Leweke, F.M.; Endepols, H. The functional networks of prepulse inhibition: Neuronal connectivity analysis based on FDG-PET in awake and unrestrained rats. Front. Behav. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Caine, S.B.; Braff, D.L.; Geyer, M.A. The neural substrates of sensorimotor gating of the startle reflex: A review of recent findings and their implications. J. Psychopharmacol. 1992, 6, 176–190. [Google Scholar] [CrossRef]
- Favalli, G.; Li, J.; Belmonte-de-Abreu, P.; Wong, A.H.; Daskalakis, Z.J. The role of BDNF in the pathophysiology and treatment of schizophrenia. J. Psychiatr. Res. 2012, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.; Wilson, C.; Terry, A.V.; Pillai, A., Jr. An inverse relationship between cortisol and BDNF levels in schizophrenia: Data from human postmortem and animal studies. Neurobiol. Dis. 2010, 39, 327–333. [Google Scholar] [CrossRef]
- Thompson Ray, M.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 2011, 36, 195–203. [Google Scholar] [CrossRef]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef]
- San-Martin, R.; Castro, L.A.; Menezes, P.R.; Fraga, F.J.; Simoes, P.W.; Salum, C. Meta-analysis of sensorimotor gating deficits in patients with schizophrenia evaluated by prepulse inhibition test. Schizophr. Bull. 2020, 46, 1482–1497. [Google Scholar] [CrossRef]
- Jaehne, E.J.; Chong, E.M.S.; Sbisa, A.; Gillespie, B.; Hill, R.; Gogos, A.; van den Buuse, M. TrkB agonist 7,8-dihydroxyflavone reverses an induced prepulse inhibition deficit selectively in maternal immune activation offspring: Implications for schizophrenia. Behav. Pharmacol. 2021, 32, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Hill, R.A. 7,8-Dihydroxyflavone as a pro-neurotrophic treatment for neurodevelopmental disorders. Neurochem. Int. 2015, 89, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wu, Y.C.; Hill, R. BDNF–TrkB signaling as a therapeutic target in neuropsychiatric disorders. J. Recep. Ligand Channel Res. 2014, 7, 61–79. [Google Scholar] [CrossRef]
- Harb, M.; Jagusch, J.; Durairaja, A.; Endres, T.; Lessmann, V.; Fendt, M. BDNF haploinsufficiency induces behavioral endophenotypes of schizophrenia in male mice that are rescued by enriched environment. Transl. Psychiatry 2021, 11, 233. [Google Scholar] [CrossRef]
- Firth, J.; Cotter, J.; Carney, R.; Yung, A.R. The pro-cognitive mechanisms of physical exercise in people with schizophrenia. Br. J. Pharmacol. 2017, 174, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Kimhy, D.; Vakhrusheva, J.; Bartels, M.N.; Armstrong, H.F.; Ballon, J.S.; Khan, S.; Chang, R.W.; Hansen, M.C.; Ayanruoh, L.; Lister, A.; et al. The impact of aerobic exercise on brain-derived neurotrophic factor and neurocognition in individuals with schizophrenia: A single-blind, randomized clinical trial. Schizophr. Bull. 2015, 41, 859–868. [Google Scholar] [CrossRef]
- Morais, A.P.D.; Pita, I.R.; Fontes-Ribeiro, C.A.; Pereira, F.C. The neurobiological mechanisms of physical exercise in methamphetamine addiction. CNS Neurosci. Ther. 2018, 24, 85–97. [Google Scholar] [CrossRef]
- Rogers, D.C.; Fisher, E.M.; Brown, S.D.; Peters, J.; Hunter, A.J.; Martin, J.E. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm. Genome 1997, 8, 711–713. [Google Scholar] [CrossRef]
- Moraga-Amaro, R.; Gonzalez, H.; Pacheco, R.; Stehberg, J. Dopamine receptor D3 deficiency results in chronic depression and anxiety. Behav. Brain Res. 2014, 274, 186–193. [Google Scholar] [CrossRef]
- Steiner, H.; Fuchs, S.; Accili, D. D3 dopamine receptor-deficient mouse: Evidence for reduced anxiety. Physiol. Behav. 1997, 63, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Chourbaji, S.; Brandwein, C.; Vogt, M.A.; Dormann, C.; Mueller, R.; Drescher, K.U.; Gross, G.; Gass, P. Dopamine receptor 3 (D3) knockout mice show regular emotional behaviour. Pharmacol. Res. 2008, 58, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kernie, S.G.; Liebl, D.J.; Parada, L.F. BDNF regulates eating behavior and locomotor activity in mice. Embo J. 2000, 19, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Davidson, C.; Lee, T.H.; Ellinwood, E.H. Acute and chronic continuous methamphetamine have different long-term behavioral and neurochemical consequences. Neurochem. Int. 2005, 46, 189–203. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chronic Saline | Chronic METH | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | WT | HET | D3KO | DM | WT | HET | D3KO | DM | |
| Male | 12 | 13 | 10 | 13 | 11 | 14 | 10 | 12 | |
| Female | 12 | 15 | 13 | 14 | 13 | 11 | 10 | 13 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hogarth, S.; Jaehne, E.J.; Xu, X.; Schwarz, Q.; van den Buuse, M. Interaction of Brain-Derived Neurotrophic Factor with the Effects of Chronic Methamphetamine on Prepulse Inhibition in Mice Is Independent of Dopamine D3 Receptors. Biomedicines 2023, 11, 2290. https://doi.org/10.3390/biomedicines11082290
Hogarth S, Jaehne EJ, Xu X, Schwarz Q, van den Buuse M. Interaction of Brain-Derived Neurotrophic Factor with the Effects of Chronic Methamphetamine on Prepulse Inhibition in Mice Is Independent of Dopamine D3 Receptors. Biomedicines. 2023; 11(8):2290. https://doi.org/10.3390/biomedicines11082290
Chicago/Turabian StyleHogarth, Samuel, Emily J. Jaehne, Xiangjun Xu, Quenten Schwarz, and Maarten van den Buuse. 2023. "Interaction of Brain-Derived Neurotrophic Factor with the Effects of Chronic Methamphetamine on Prepulse Inhibition in Mice Is Independent of Dopamine D3 Receptors" Biomedicines 11, no. 8: 2290. https://doi.org/10.3390/biomedicines11082290
APA StyleHogarth, S., Jaehne, E. J., Xu, X., Schwarz, Q., & van den Buuse, M. (2023). Interaction of Brain-Derived Neurotrophic Factor with the Effects of Chronic Methamphetamine on Prepulse Inhibition in Mice Is Independent of Dopamine D3 Receptors. Biomedicines, 11(8), 2290. https://doi.org/10.3390/biomedicines11082290