
Does β-Hydroxy-β-Methylbutyrate Have Any Potential to Support the Treatment of Duchenne Muscular Dystrophy in Humans and Animals?




Abstract
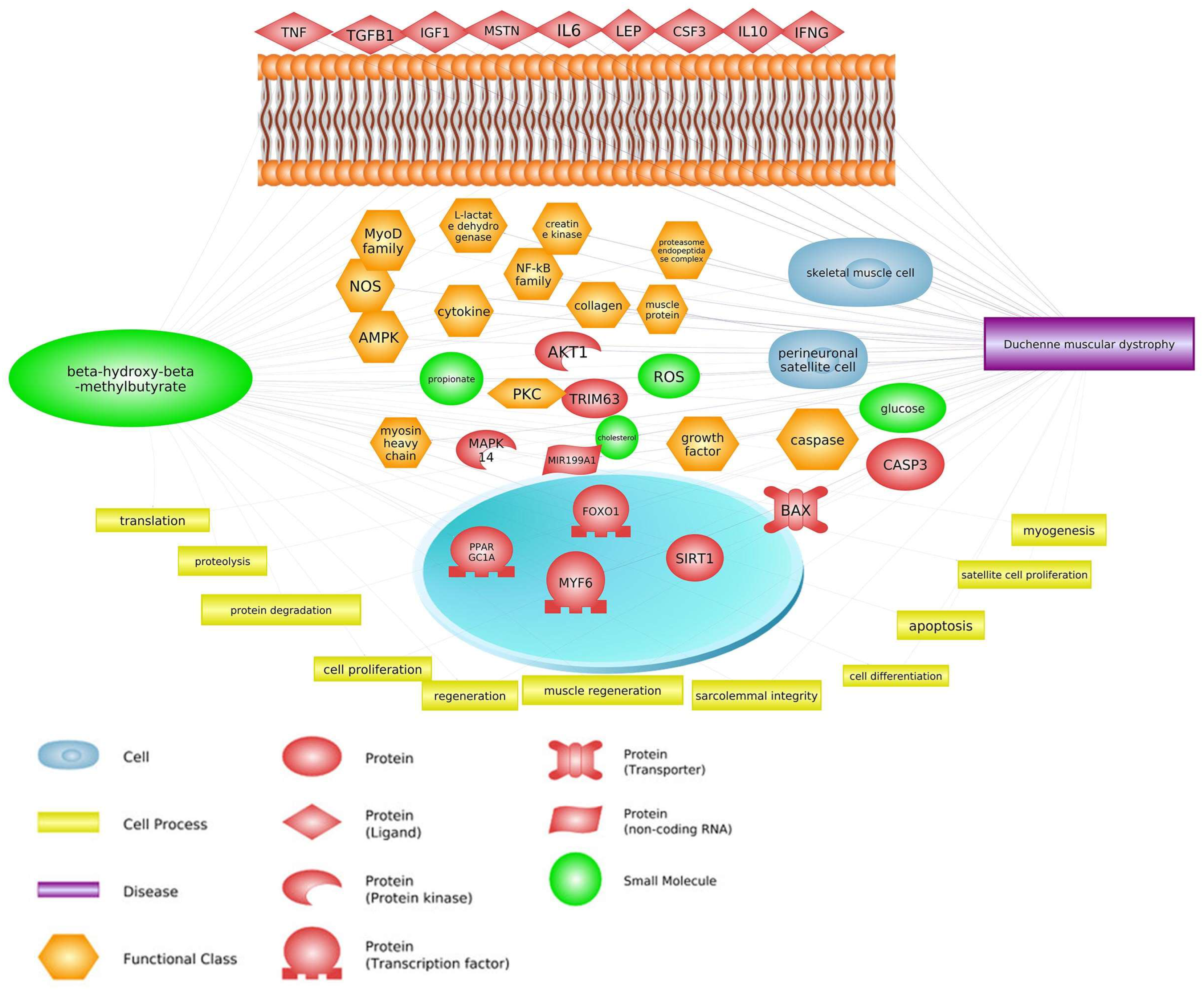
:1. Introduction
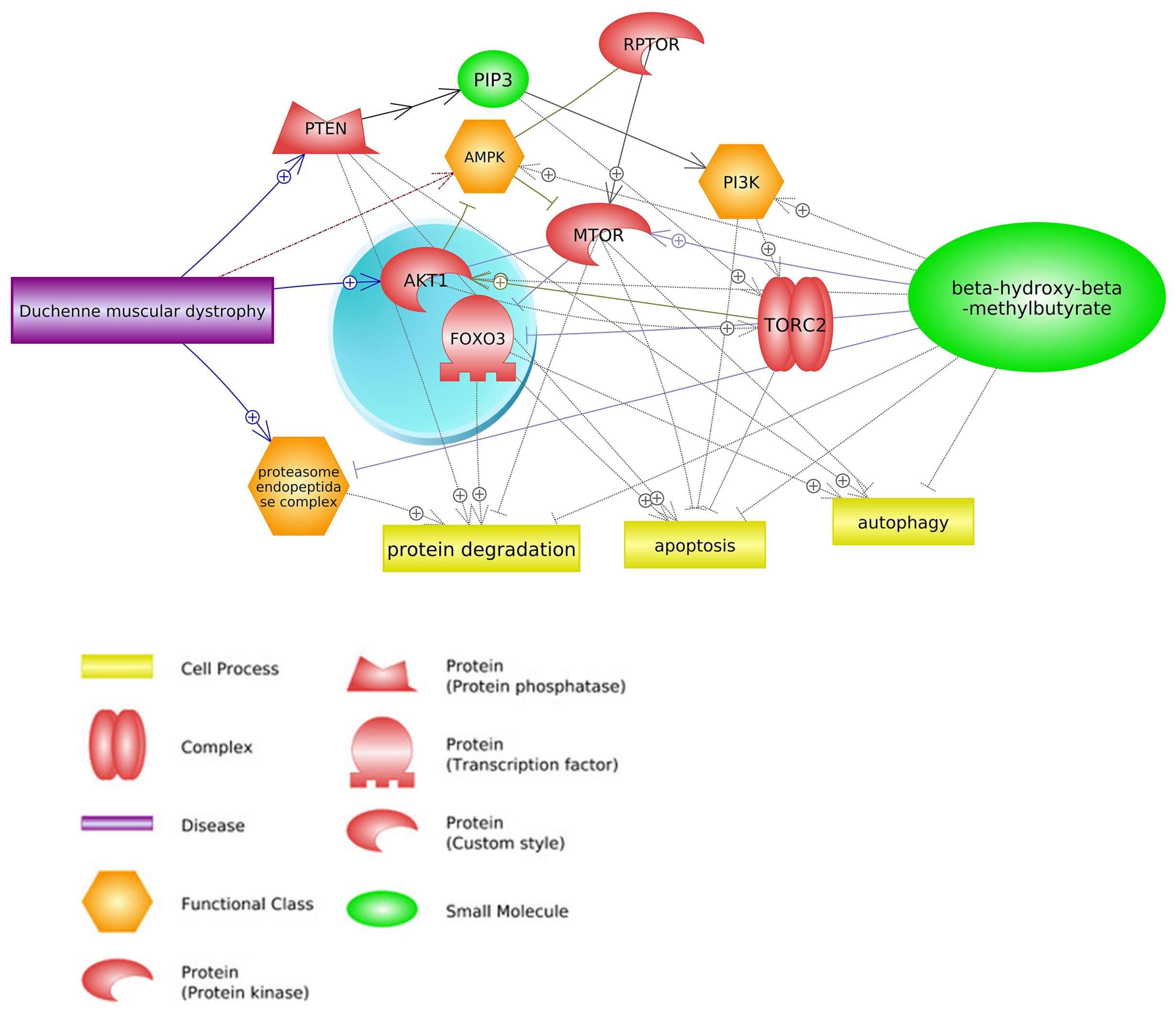
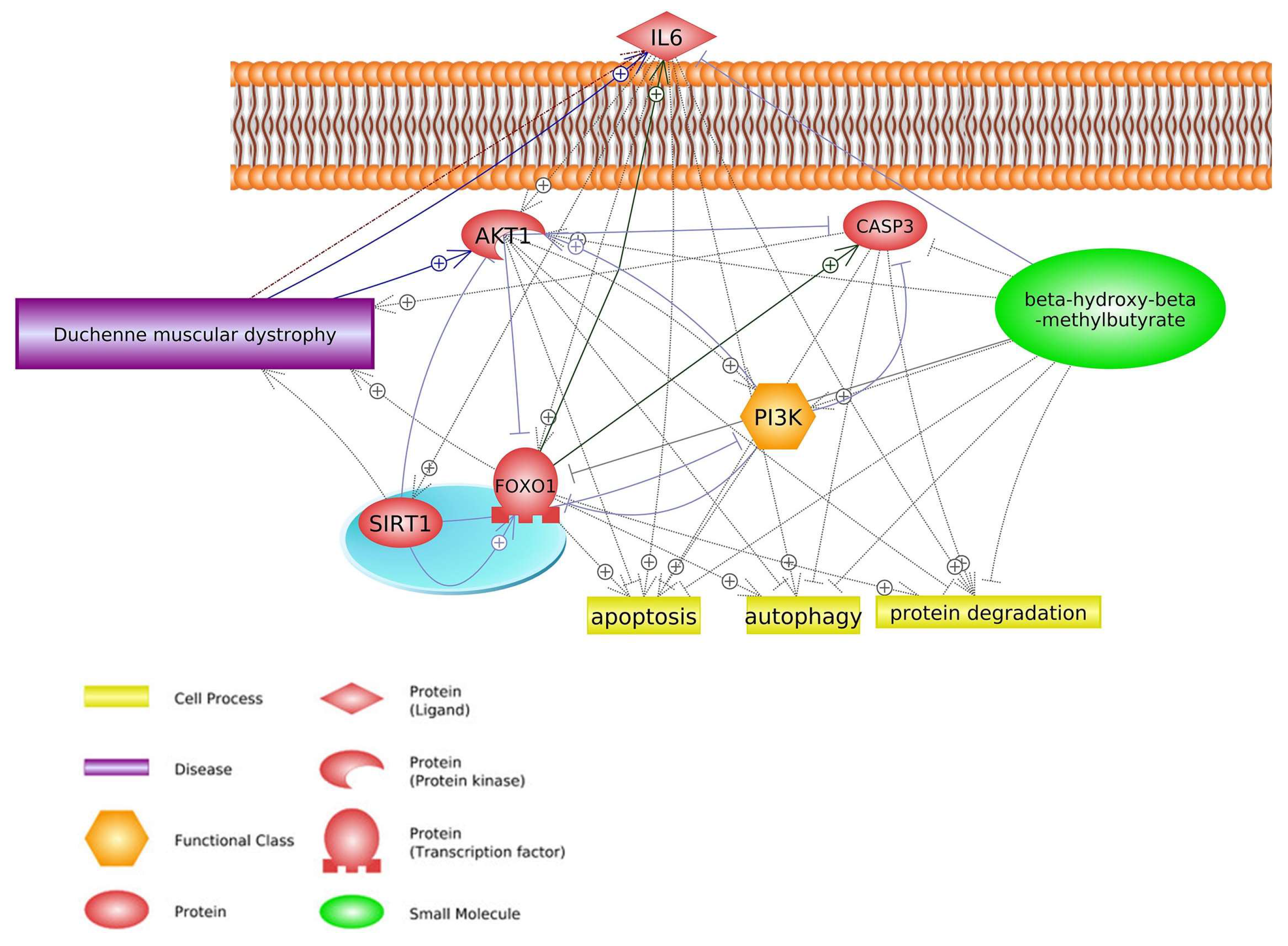
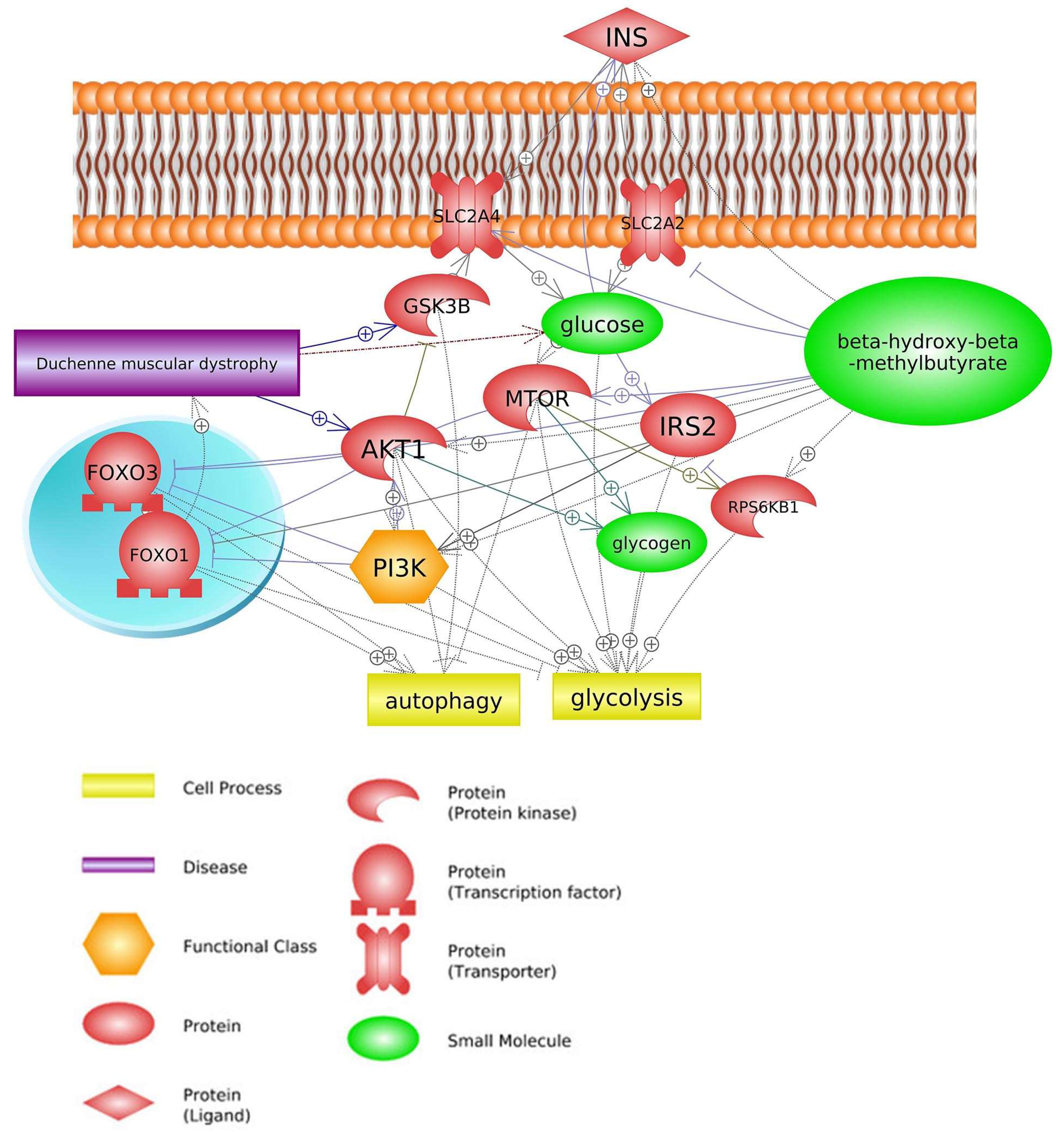
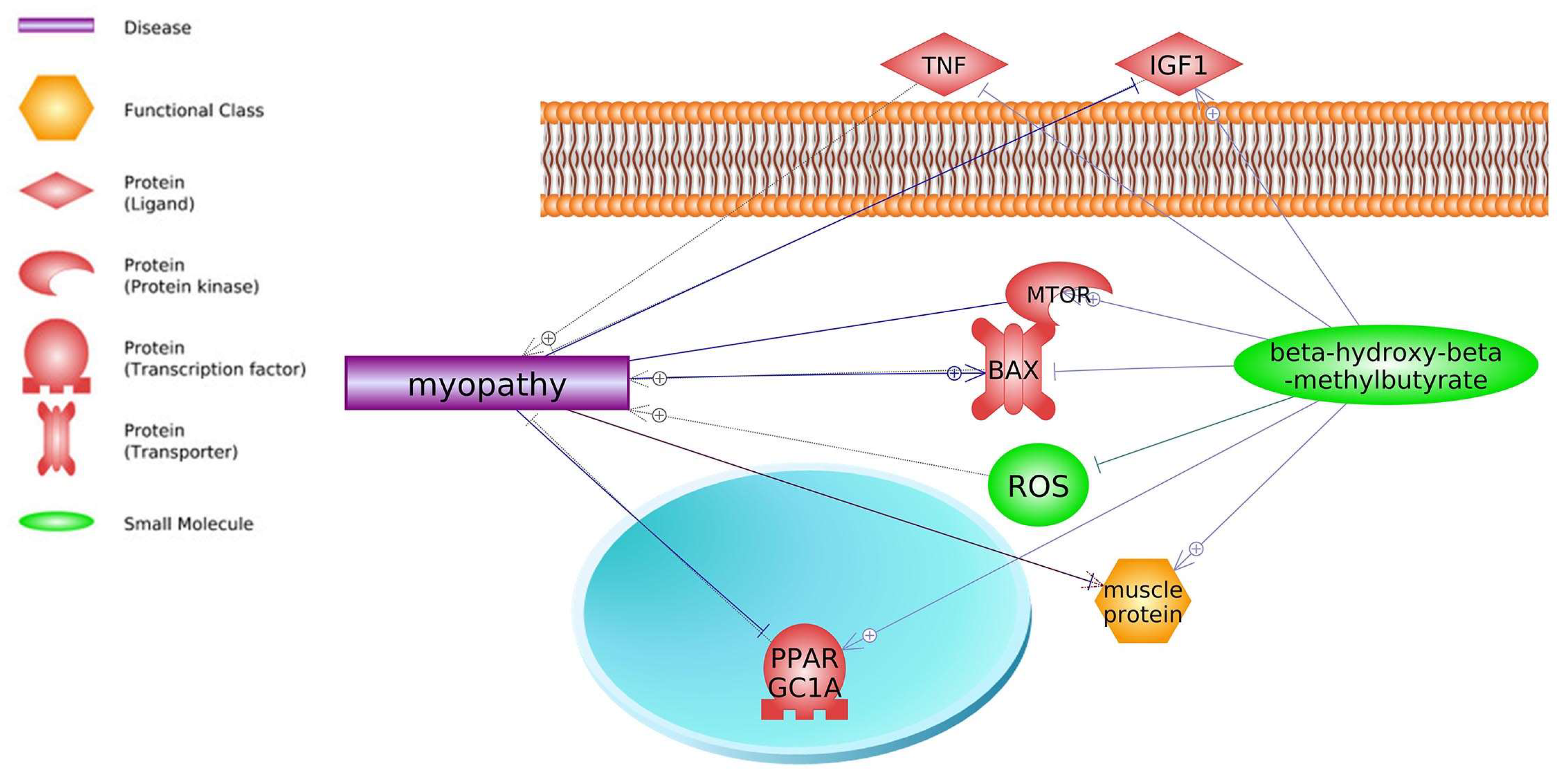
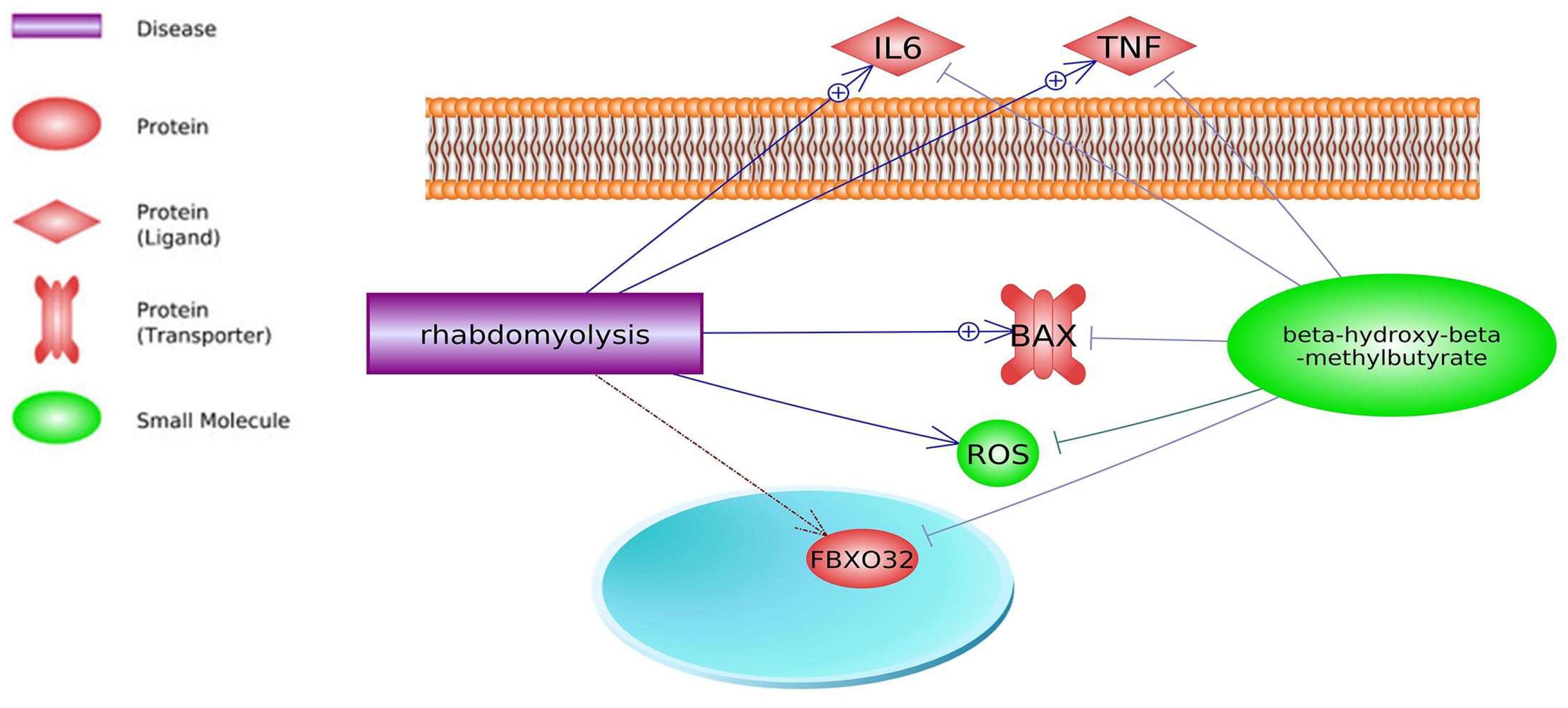
2. Common Pathways
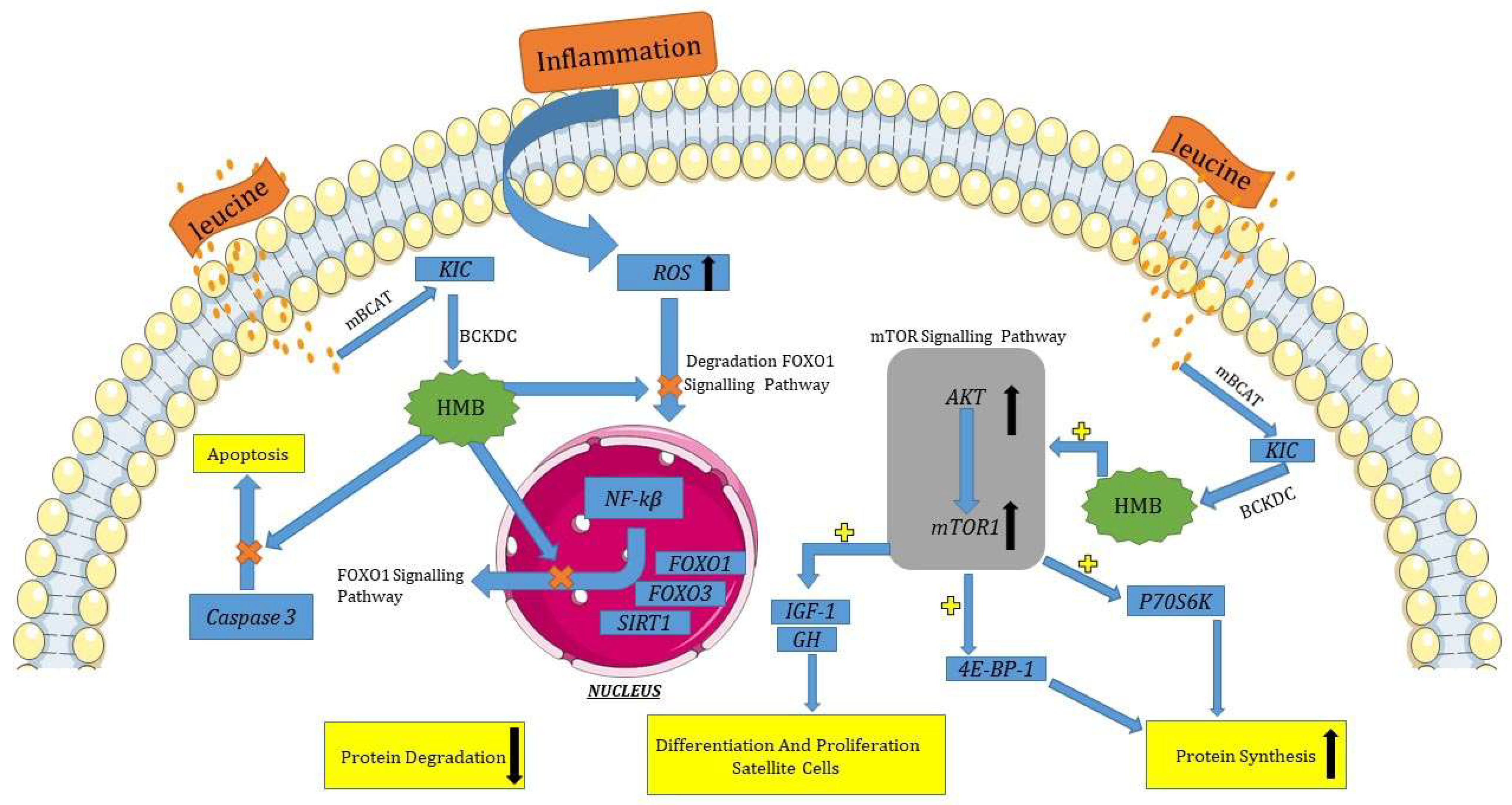
2.1. mTOR Signaling Pathway

2.2. FOXO1 Signaling Pathway

2.3. Insulin Signaling Pathway

3. Common Genes
4. Therapeutic Agents Used for DMD Treatment
4.1. Statins


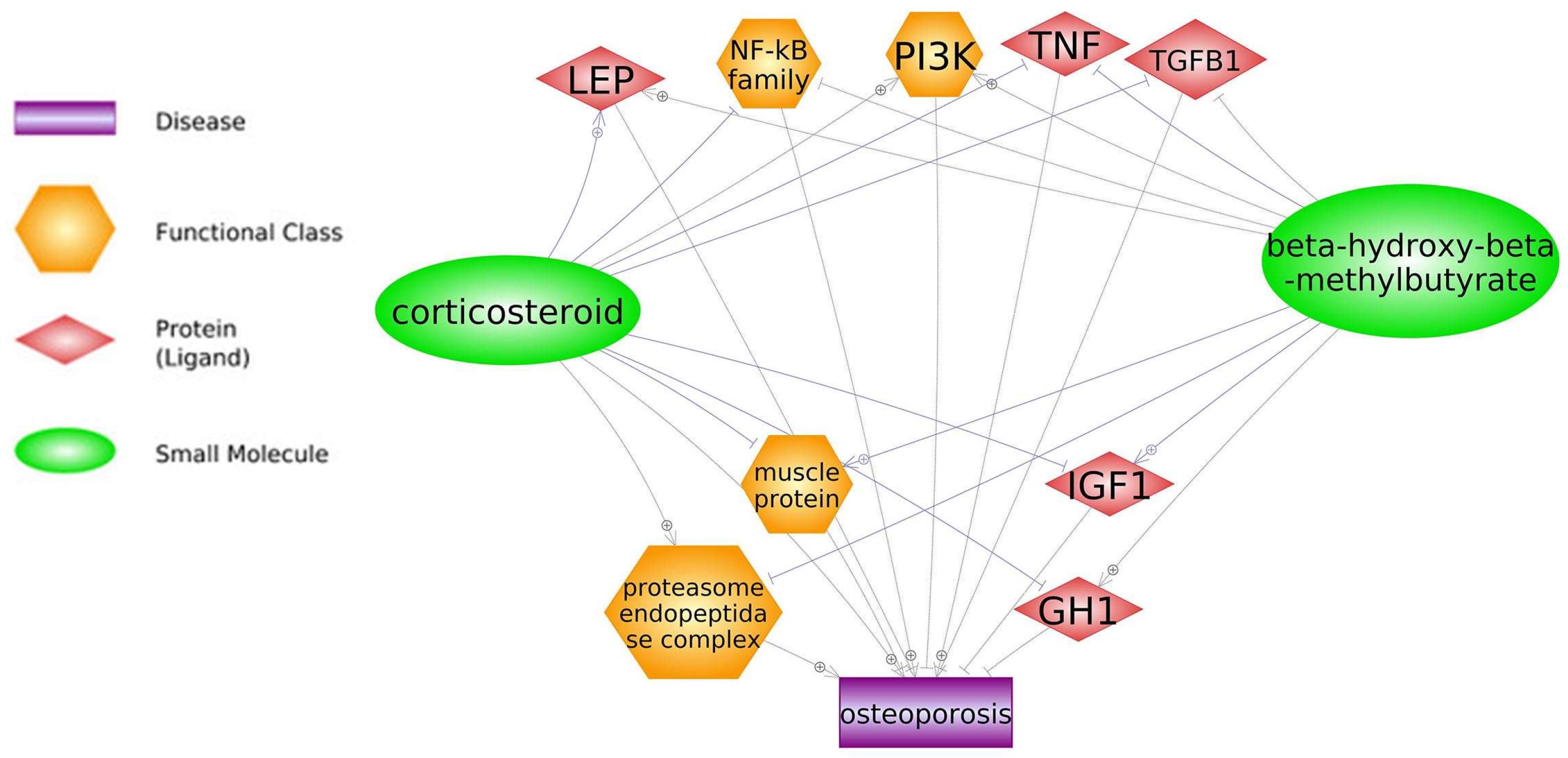
4.2. Glucocorticoids

5. Conclusions

Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Nissen, S.L.; Abumrad, N.N. Nutritional Role of the Leucine Metabolite β-Hydroxy β-Methylbutyrate (HMB). J. Nutr. Biochem. 1997, 8, 300–311. [Google Scholar] [CrossRef]
- Engelen, M.P.K.J.; Deutz, N.E.P. Is HMB an Effective Anabolic Agent to Improve Outcome in Older Diseased Populations? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.J.; Hossain, T.; Limb, M.C.; Phillips, B.E.; Lund, J.; Williams, J.P.; Brook, M.S.; Cegielski, J.; Philp, A.; Ashcroft, S.; et al. Impact of the Calcium Form of β-Hydroxy-β-Methylbutyrate upon Human Skeletal Muscle Protein Metabolism. Clin. Nutr. Edinb. Scotl. 2018, 37, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Khal, J.; Tisdale, M.J. Downregulation of Ubiquitin-Dependent Protein Degradation in Murine Myotubes during Hyperthermia by Eicosapentaenoic Acid. Biochem. Biophys. Res. Commun. 2005, 332, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M.; Muthny, T.; Kovarik, M.; Sispera, L. Effect of Beta-Hydroxy-Beta-Methylbutyrate (HMB) on Protein Metabolism in Whole Body and in Selected Tissues. Food Chem. Toxicol. 2009, 47, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.H.; Feleke, G.; Din, M.; Yasmin, T.; Singh, G.; Khan, F.A.; Rathmacher, J.A. Nutritional Treatment for Acquired Immunodeficiency Virus-Associated Wasting Using β-Hydroxy β-Methylbutyrate, Glutamine, and Arginine: A Randomized, Double-Blind, Placebo-Controlled Study. J. Parenter. Enter. Nutr. 2000, 24, 133–139. [Google Scholar] [CrossRef]
- Kuhls, D.A.; Rathmacher, J.A.; Musngi, M.D.; Frisch, D.A.; Nielson, J.; Barber, A.; MacIntyre, A.D.; Coates, J.E.; Fildes, J.J. Beta-Hydroxy-Beta-Methylbutyrate Supplementation in Critically Ill Trauma Patients. J. Trauma 2007, 62, 125–131; discussion 131–132. [Google Scholar] [CrossRef]
- Bear, D.E.; Langan, A.; Dimidi, E.; Wandrag, L.; Harridge, S.D.; Hart, N.; Connolly, B.; Whelan, K. β-Hydroxy-β-Methylbutyrate and Its Impact on Skeletal Muscle Mass and Physical Function in Clinical Practice: A Systematic Review and Meta-Analysis. Am. J. Clin. Nutr. 2019, 109, 1119–1132. [Google Scholar] [CrossRef]
- Aversa, Z.; Bonetto, A.; Costelli, P.; Minero, V.G.; Penna, F.; Baccino, F.M.; Lucia, S.; Rossi Fanelli, F.; Muscaritoli, M. β-Hydroxy-β-Methylbutyrate (HMB) Attenuates Muscle and Body Weight Loss in Experimental Cancer Cachexia. Int. J. Oncol. 2011, 38, 713–720. [Google Scholar]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Mechanism of Attenuation of Muscle Protein Degradation Induced by Tumor Necrosis Factor-α and Angiotensin II by β-Hydroxy-β-Methylbutyrate. Am. J. Physiol.-Endocrinol. Metab. 2008, 295, E1417–E1426. [Google Scholar] [CrossRef]
- Kornasio, R.; Riederer, I.; Butler-Browne, G.; Mouly, V.; Uni, Z.; Halevy, O. β-Hydroxy-β-Methylbutyrate (HMB) Stimulates Myogenic Cell Proliferation, Differentiation and Survival via the MAPK/ERK and PI3K/Akt Pathways. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of Muscular Dystrophies: A Systematic Literature Review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Campbell, K.P. Muscular Dystrophies and the Dystrophin-Glycoprotein Complex. Curr. Opin. Neurol. 1997, 10, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Del Rocío Cruz-Guzmán, O.; Rodríguez-Cruz, M.; Escobar Cedillo, R.E. Systemic Inflammation in Duchenne Muscular Dystrophy: Association with Muscle Function and Nutritional Status. BioMed Res. Int. 2015, 2015. [Google Scholar]
- Tidball, J.G. Inflammatory Processes in Muscle Injury and Repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef]
- Rodríguez-Cruz, M.; Cruz-Guzmán, O.D.R.; Almeida-Becerril, T.; Solís-Serna, A.D.; Atilano-Miguel, S.; Sánchez-González, J.R.; Barbosa-Cortés, L.; Ruíz-Cruz, E.D.; Huicochea, J.C.; Cárdenas-Conejo, A.; et al. Potential Therapeutic Impact of Omega-3 Long Chain-Polyunsaturated Fatty Acids on Inflammation Markers in Duchenne Muscular Dystrophy: A Double-Blind, Controlled Randomized Trial. Clin. Nutr. Edinb. Scotl. 2018, 37, 1840–1851. [Google Scholar] [CrossRef]
- De Paepe, B.; De Bleecker, J.L. Cytokines and Chemokines as Regulators of Skeletal Muscle Inflammation: Presenting the Case of Duchenne Muscular Dystrophy. Mediat. Inflamm. 2013, 2013, 540370. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Guiraud, S.; Davies, K.E. Pharmacological Advances for Treatment in Duchenne Muscular Dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population Frequencies of Inherited Neuromuscular Diseases—A World Survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Lee, J.J.; Tidball, J.G. Prednisolone Decreases Cellular Adhesion Molecules Required for Inflammatory Cell Infiltration in Dystrophin-Deficient Skeletal Muscle. Neuromuscul. Disord. NMD 2004, 14, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Payne, E.T.; Yasuda, N.; Bourgeois, J.M.; Devries, M.C.; Rodriguez, M.C.; Yousuf, J.; Tarnopolsky, M.A. Nutritional Therapy Improves Function and Complements Corticosteroid Intervention in Mdx Mice. Muscle Nerve 2006, 33, 66–77. [Google Scholar] [CrossRef]
- Majewska, A.; Domoradzki, T.; Grzelkowska-Kowalczyk, K. Transcriptomic Profiling During Myogenesis. Methods Mol. Biol. 2019, 1889, 127–168. [Google Scholar] [CrossRef]
- Howell, J.J.; Manning, B.D. MTOR Couples Cellular Nutrient Sensing to Organismal Metabolic Homeostasis. Trends Endocrinol. Metab. 2011, 22, 94–102. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. MTOR Kinase Structure, Mechanism and Regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Yamamoto, M.; Teramoto, K.; Katoh, H. Epidermal Growth Factor Promotes Glioblastoma Cell Death under Glucose Deprivation via Upregulation of XCT (SLC7A11). Cell. Signal. 2021, 78, 109874. [Google Scholar] [CrossRef]
- Qian, B.; Yao, Z.; Yang, Y.; Li, N.; Wang, Q. Downregulation of SDCBP Inhibits Cell Proliferation and Induces Apoptosis by Regulating PI3K/AKT/MTOR Pathway in Gastric Carcinoma. Biotechnol. Appl. Biochem. 2022, 69, 240–247. [Google Scholar] [CrossRef]
- Pimentel, G.D.; Rosa, J.C.; Lira, F.S.; Zanchi, N.E.; Ropelle, E.R.; Oyama, L.M.; Oller do Nascimento, C.M.; de Mello, M.T.; Tufik, S.; Santos, R.V. β-Hydroxy-β-Methylbutyrate (HMβ) Supplementation Stimulates Skeletal Muscle Hypertrophy in Rats via the MTOR Pathway. Nutr. Metab. 2011, 8, 11. [Google Scholar] [CrossRef]
- Wan, H.; Zhu, J.; Su, G.; Liu, Y.; Hua, L.; Hu, L.; Wu, C.; Zhang, R.; Zhou, P.; Shen, Y.; et al. Dietary Supplementation with β-Hydroxy-β-Methylbutyrate Calcium during the Early Postnatal Period Accelerates Skeletal Muscle Fibre Growth and Maturity in Intra-Uterine Growth-Retarded and Normal-Birth-Weight Piglets. Br. J. Nutr. 2016, 115, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- de, A. Costa Riela, N.; Alvim Guimarães, M.M.; Oliveira de Almeida, D.; Araujo, E.M.Q. Effects of Beta-Hydroxy-Beta-Methylbutyrate Supplementation on Elderly Body Composition and Muscle Strength: A Review of Clinical Trials. Ann. Nutr. Metab. 2021, 77, 16–22. [Google Scholar] [CrossRef]
- Baier, S.; Johannsen, D.; Abumrad, N.; Rathmacher, J.A.; Nissen, S.; Flakoll, P. Year-Long Changes in Protein Metabolism in Elderly Men and Women Supplemented with a Nutrition Cocktail of β-Hydroxy-β-Methylbutyrate (HMB), L-Arginine, and L-Lysine. J. Parenter. Enter. Nutr. 2009, 33, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.R.; Fukuda, D.H.; Kendall, K.L.; Smith-Ryan, A.E.; Moon, J.R.; Hoffman, J.R. β-Hydroxy-β-Methylbutyrate (HMB) Supplementation and Resistance Exercise Significantly Reduce Abdominal Adiposity in Healthy Elderly Men. Exp. Gerontol. 2015, 64, 33–34. [Google Scholar] [CrossRef]
- Ellis, A.C.; Hunter, G.R.; Goss, A.M.; Gower, B.A. Oral Supplementation with Beta-Hydroxy-Beta-Methylbutyrate, Arginine, and Glutamine Improves Lean Body Mass in Healthy Older Adults. J. Diet. Suppl. 2019, 16, 281–293. [Google Scholar] [CrossRef]
- Ortiz, A. β-Hydroxy-β-Methylbutyrate Supplementation in Special Populations. Strength Cond. J. 2013, 35, 73–77. [Google Scholar] [CrossRef]
- Portal, S.; Zadik, Z.; Rabinowitz, J.; Pilz-Burstein, R.; Adler-Portal, D.; Meckel, Y.; Cooper, D.M.; Eliakim, A.; Nemet, D. The Effect of HMB Supplementation on Body Composition, Fitness, Hormonal and Inflammatory Mediators in Elite Adolescent Volleyball Players: A Prospective Randomized, Double-Blind, Placebo-Controlled Study. Eur. J. Appl. Physiol. 2011, 111, 2261–2269. [Google Scholar] [CrossRef]
- Amini-Khoei, H.; Saghaei, E.; Mobini, G.-R.; Sabzevary-Ghahfarokhi, M.; Ahmadi, R.; Bagheri, N.; Mokhtari, T. Possible Involvement of PI3K/AKT/MTOR Signaling Pathway in the Protective Effect of Selegiline (Deprenyl) against Memory Impairment Following Ischemia Reperfusion in Rat. Neuropeptides 2019, 77, 101942. [Google Scholar] [CrossRef]
- Brooks, D.L.; Garza, A.E.; Katayama, I.A.; Romero, J.R.; Adler, G.K.; Pojoga, L.H.; Williams, G.H. Aldosterone Modulates the Mechanistic Target of Rapamycin Signaling in Male Mice. Endocrinology 2019, 160, 716–728. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of MTOR Complexes: MTORC1 and MTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Kumari, S.; Khan, S.; Sekhri, R.; Mandil, H.; Behrman, S.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Protein Kinase D1 Regulates Metabolic Switch in Pancreatic Cancer via Modulation of MTORC1. Br. J. Cancer 2020, 122, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L. Insulin Antagonizes Ischemia-Induced Thr172 Phosphorylation of AMP-Activated Protein Kinase α-Subunits in Heart via Hierarchical Phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730. [Google Scholar] [CrossRef] [PubMed]
- Fitschen, P.J.; Wilson, G.J.; Wilson, J.M.; Wilund, K.R. Efficacy of β-Hydroxy-β-Methylbutyrate Supplementation in Elderly and Clinical Populations. Nutrition 2013, 29, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; V Khamoui, A.; Jo, E.; Park, B.-S.; Jun Lee, W. β-Hydroxy-β-Methylbutyrate as a Countermeasure for Cancer Cachexia: A Cellular and Molecular Rationale. Anti-Cancer Agents Med. Chem. 2013, 13, 1188–1196. [Google Scholar] [CrossRef]
- Peter, A.K.; Crosbie, R.H. Hypertrophic Response of Duchenne and Limb-Girdle Muscular Dystrophies Is Associated with Activation of Akt Pathway. Exp. Cell Res. 2006, 312, 2580–2591. [Google Scholar] [CrossRef]
- Alway, S.E.; Pereira, S.L.; Edens, N.K.; Hao, Y.; Bennett, B.T. β-Hydroxy-β-Methylbutyrate (HMB) Enhances the Proliferation of Satellite Cells in Fast Muscles of Aged Rats during Recovery from Disuse Atrophy. Exp. Gerontol. 2013, 48, 973–984. [Google Scholar] [CrossRef]
- Salto, R.; Vílchez, J.D.; Girón, M.D.; Cabrera, E.; Campos, N.; Manzano, M.; Rueda, R.; López-Pedrosa, J.M. β-Hydroxy-β-Methylbutyrate (HMB) Promotes Neurite Outgrowth in Neuro2a Cells. PLoS ONE 2015, 10, e0135614. [Google Scholar] [CrossRef]
- Zhong, Y.; Zeng, L.; Deng, J.; Duan, Y.; Li, F. β-Hydroxy-β-Methylbutyrate (HMB) Improves Mitochondrial Function in Myocytes through Pathways Involving PPARβ/δ and CDK4. Nutrition 2019, 60, 217–226. [Google Scholar] [CrossRef]
- Parveen, A.; Wen, Y.; Roy, A.; Kumar, A. Therapeutic Targeting of PTEN in Duchenne Muscular Dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zheng, C.; Song, B.; Guo, Q.; Zhong, Y.; Zhang, S.; Zhang, L.; Duan, G.; Li, F.; Duan, Y. HMB Improves Lipid Metabolism of Bama Xiang Mini-Pigs via Modulating the Bacteroidetes-Acetic Acid-AMPKα Axis. Front. Microbiol. 2021, 12, 736997. [Google Scholar] [CrossRef] [PubMed]
- Wattin, M.; Gaweda, L.; Muller, P.; Baritaud, M.; Scholtes, C.; Lozano, C.; Gieseler, K.; Kretz-Remy, C. Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2018, 19, 178. [Google Scholar] [CrossRef] [PubMed]
- Mirza, K.A.; Pereira, S.L.; Voss, A.C.; Tisdale, M.J. Comparison of the Anticatabolic Effects of Leucine and Ca-β-Hydroxy-β-Methylbutyrate in Experimental Models of Cancer Cachexia. Nutrition 2014, 30, 807–813. [Google Scholar] [CrossRef]
- Gross, D.N.; Van Den Heuvel, A.P.J.; Birnbaum, M.J. The Role of FoxO in the Regulation of Metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO Transcription Factors at the Interface between Longevity and Tumor Suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Zubair, A.; Frieri, M. Role of Nuclear Factor-ĸB in Breast and Colorectal Cancer. Curr. Allergy Asthma Rep. 2013, 13, 44–49. [Google Scholar] [CrossRef]
- Miyake, S.; Ogo, A.; Kubota, H.; Teramoto, F.; Hirai, T. β-Hydroxy-β-Methylbutyrate Suppresses NF-\sckB Activation and IL6 Production in TE-1 Cancer Cells. In Vivo 2019, 33, 353–358. [Google Scholar] [CrossRef]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-KappaB Pathway in Duchenne Muscular Dystrophy: Relation to Age. Acta Myol. 2011, 30, 16–23. [Google Scholar]
- Gallardo, F.S.; Córdova-Casanova, A.; Brandan, E. The Linkage between Inflammation and Fibrosis in Muscular Dystrophies: The Axis Autotaxin–Lysophosphatidic Acid as a New Therapeutic Target? J. Cell Commun. Signal. 2021, 15, 317–334. [Google Scholar] [CrossRef]
- Dudek, M.; Lohr, K.; Donakonda, S.; Baumann, T.; Lüdemann, M.; Hegenbarth, S.; Duebbel, L.; Eberhagen, C.; Michailidou, S.; Yassin, A. IL6-Induced FOXO1 Activity Determines the Dynamics of Metabolism in CD8 T Cells Cross-Primed by Liver Sinusoidal Endothelial Cells. Cell Rep. 2022, 38, 110389. [Google Scholar] [CrossRef] [PubMed]
- Ait Ahmed, Y.; Fu, Y.; Rodrigues, R.M.; He, Y.; Guan, Y.; Guillot, A.; Ren, R.; Feng, D.; Hidalgo, J.; Ju, C. Kupffer Cell Restoration after Partial Hepatectomy Is Mainly Driven by Local Cell Proliferation in IL6-Dependent Autocrine and Paracrine Manners. Cell. Mol. Immunol. 2021, 18, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; El Meslemani, A.H.; Sandri, C.; Schjerling, P.; Vissing, K.; Andersen, J.L.; Rossini, K.; Carraro, U.; Angelini, C. Caspase 3 Expression Correlates with Skeletal Muscle Apoptosis in Duchenne and Facioscapulo Human Muscular Dystrophy. A Potential Target for Pharmacological Treatment? J. Neuropathol. Exp. Neurol. 2001, 60, 302–312. [Google Scholar] [CrossRef]
- Almushayt, S.J.; Hussain, S.; Wilkinson, D.J.; Selby, N.M. A Systematic Review of the Acute Effects of Hemodialysis on Skeletal Muscle Perfusion, Metabolism, and Function. Kidney Int. Rep. 2020, 5, 307–317. [Google Scholar] [CrossRef]
- Noh, K.K.; Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Park, C.H.; Yoon, C.; Kim, N.D.; Kim, M.K.; Chung, H.Y. β-Hydroxy β-Methylbutyrate Improves Dexamethasone-Induced Muscle Atrophy by Modulating the Muscle Degradation Pathway in SD Rat. PLoS ONE 2014, 9, e102947. [Google Scholar] [CrossRef]
- Hao, Y.; Jackson, J.R.; Wang, Y.; Edens, N.; Pereira, S.L.; Alway, S.E. β-Hydroxy-β-Methylbutyrate Reduces Myonuclear Apoptosis during Recovery from Hind Limb Suspension-Induced Muscle Fiber Atrophy in Aged Rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R701–R715. [Google Scholar] [CrossRef]
- Alexander, M.S.; Casar, J.C.; Motohashi, N.; Vieira, N.M.; Eisenberg, I.; Marshall, J.L.; Gasperini, M.J.; Lek, A.; Myers, J.A.; Estrella, E.A. MicroRNA-486–Dependent Modulation of DOCK3/PTEN/AKT Signaling Pathways Improves Muscular Dystrophy–Associated Symptoms. J. Clin. Investig. 2014, 124, 2651–2667. [Google Scholar] [CrossRef]
- Yao, C.; Guo, G.; Huang, R.; Tang, C.; Zhu, Q.; Cheng, Y.; Kong, L.; Ren, J.; Fang, M. Manual Therapy Regulates Oxidative Stress in Aging Rat Lumbar Intervertebral Discs through the SIRT1/FOXO1 Pathway. Aging 2022, 14, 2400. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic Beta Cells Creates an Insulin Secretory Defect Similar to That in Type 2 Diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Vickers, N.J. Animal Communication: When I’m Calling You, Will You Answer Too? Curr. Biol. 2017, 27, R713–R715. [Google Scholar] [CrossRef] [PubMed]
- Shaham, O.; Wei, R.; Wang, T.J.; Ricciardi, C.; Lewis, G.D.; Vasan, R.S.; Carr, S.A.; Thadhani, R.; Gerszten, R.E.; Mootha, V.K. Metabolic Profiling of the Human Response to a Glucose Challenge Reveals Distinct Axes of Insulin Sensitivity. Mol. Syst. Biol. 2008, 4, 214. [Google Scholar] [CrossRef]
- Sharma, U.; Atri, S.; Sharma, M.C.; Sarkar, C.; Jagannathan, N.R. Skeletal Muscle Metabolism in Duchenne Muscular Dystrophy (DMD): An in-Vitro Proton NMR Spectroscopy Study. Magn. Reson. Imaging 2003, 21, 145–153. [Google Scholar] [CrossRef]
- Schneider, S.M.; Sridhar, V.; Bettis, A.K.; Heath-Barnett, H.; Balog-Alvarez, C.J.; Guo, L.-J.; Johnson, R.; Jaques, S.; Vitha, S.; Glowcwski, A.C. Glucose Metabolism as a Pre-Clinical Biomarker for the Golden Retriever Model of Duchenne Muscular Dystrophy. Mol. Imaging Biol. 2018, 20, 780–788. [Google Scholar] [CrossRef]
- Proto, J.D.; Tang, Y.; Lu, A.; Chen, W.C.W.; Stahl, E.; Poddar, M.; Beckman, S.A.; Robbins, P.D.; Nidernhofer, L.J.; Imbrogno, K. NF-ΚB Inhibition Reveals a Novel Role for HGF during Skeletal Muscle Repair. Cell Death Dis. 2015, 6, e1730. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Hossain, T.; Hill, D.S.; Phillips, B.E.; Crossland, H.; Williams, J.; Loughna, P.; Churchward-Venne, T.A.; Breen, L.; Phillips, S.M.; et al. Effects of Leucine and Its Metabolite β-Hydroxy-β-Methylbutyrate on Human Skeletal Muscle Protein Metabolism. J. Physiol. 2013, 591, 2911–2923. [Google Scholar] [CrossRef]
- Schnuck, J.K.; Johnson, M.A.; Gould, L.M.; Gannon, N.P.; Vaughan, R.A. Acute β-Hydroxy-β-Methyl Butyrate Suppresses Regulators of Mitochondrial Biogenesis and Lipid Oxidation While Increasing Lipid Content in Myotubes. Lipids 2016, 51, 1127–1136. [Google Scholar] [CrossRef]
- Sharawy, M.H.; El-Awady, M.S.; Megahed, N.; Gameil, N.M. The Ergogenic Supplement β-Hydroxy-β-Methylbutyrate (HMB) Attenuates Insulin Resistance through Suppressing GLUT-2 in Rat Liver. Can. J. Physiol. Pharmacol. 2016, 94, 488–497. [Google Scholar] [CrossRef]
- Chiu, W.; Hsun, Y.-H.; Chang, K.-J.; Yarmishyn, A.A.; Hsiao, Y.-J.; Chien, Y.; Chien, C.-S.; Ma, C.; Yang, Y.-P.; Tsai, P.-H. Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. Int. J. Mol. Sci. 2020, 21, 9589. [Google Scholar] [CrossRef] [PubMed]
- Shirvani, H.; Rahmati-Ahmadabad, S.; Kowsari, E.; Fry, H.; Kazemi, M.; Kaviani, M. Effects of 2-Week HMB-FA Supplementation with or without Eccentric Resistance Exercise on Expression of Some Genes Related to Muscle Protein Turnover and Serum Irisin and IGF1 IGF1 Concentrations. Gene 2020, 760, 145018. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Childers, M.K.; Bogan, D.J.; Bogan, J.R.; Nghiem, P.; Wang, J.; Fan, Z.; Howard, J.F.; Schatzberg, S.J.; Dow, J.L.; et al. The Paradox of Muscle Hypertrophy in Muscular Dystrophy. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 149–172. [Google Scholar] [CrossRef] [PubMed]
- Söderpalm, A.-C.; Magnusson, P.; Åhlander, A.-C.; Karlsson, J.; Kroksmark, A.-K.; Tulinius, M.; Swolin-Eide, D. Low Bone Mineral Density and Decreased Bone Turnover in Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2007, 17, 919–928. [Google Scholar] [CrossRef]
- Świetlicka, I.; Muszyński, S.; Prein, C.; Clausen-Schaumann, H.; Aszodi, A.; Arciszewski, M.B.; Blicharski, T.; Gagoś, M.; Świetlicki, M.; Dobrowolski, P.; et al. Fourier Transform Infrared Microspectroscopy Combined with Principal Component Analysis and Artificial Neural Networks for the Study of the Effect of β-Hydroxy-β-Methylbutyrate (HMB) Supplementation on Articular Cartilage. Int. J. Mol. Sci. 2021, 22, 9189. [Google Scholar] [CrossRef]
- Cotán, D.; Villanueva Paz, M.; Alcocer-Gómez, E.; Garrido-Maraver, J.; de la Mata, M.; Delgado Pavón, A.; de Lavera, I.; Galán, F.; Ybot-González, P.; A Sánchez-Alcázar, J. AMPK as a Target in Rare Diseases. Curr. Drug Targets 2016, 17, 921–931. [Google Scholar] [CrossRef]
- Gonçalves, M.A.; Janssen, J.M.; Nguyen, Q.G.; Athanasopoulos, T.; Hauschka, S.D.; Dickson, G.; De Vries, A.A. Transcription Factor Rational Design Improves Directed Differentiation of Human Mesenchymal Stem Cells Into Skeletal Myocytes. Mol. Ther. 2011, 19, 1331–1341. [Google Scholar] [CrossRef]
- Gonçalves, M.A.; Swildens, J.; Holkers, M.; Narain, A.; Van Nierop, G.P.; Van De Watering, M.J.; Knaän-Shanzer, S.; De Vries, A.A. Genetic Complementation of Human Muscle Cells via Directed Stem Cell Fusion. Mol. Ther. 2008, 16, 741–748. [Google Scholar] [CrossRef]
- Tian, L.J.; Cao, J.H.; Deng, X.Q.; Zhang, C.L.; Qian, T.; Song, X.X.; Huang, B.S. Gene Expression Profiling of Duchenne Muscular Dystrophy Reveals Characteristics along Disease Progression. Genet. Mol. Res. 2014, 13, 1402–1411. [Google Scholar] [CrossRef]
- Peterson, A.L.; Qureshi, M.A.; Ferket, P.R.; Jr, J.C.F. In Vitro Exposure with β-Hydroxy-β-Methylbutyrate Enhances Chicken Macrophage Growth and Function. Vet. Immunol. Immunopathol. 1999, 67, 67–78. [Google Scholar] [CrossRef]
- Finkel, R.S.; Finanger, E.; Vandenborne, K.; Sweeney, H.L.; Tennekoon, G.; Shieh, P.B.; Willcocks, R.; Walter, G.; Rooney, W.D.; Forbes, S.C. Disease-Modifying Effects of Edasalonexent, an NF-ΚB Inhibitor, in Young Boys with Duchenne Muscular Dystrophy: Results of the MoveDMD Phase 2 and Open Label Extension Trial. Neuromuscul. Disord. 2021, 31, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Luce, L.N.; Dalamon, V.; Ferrer, M.; Parma, D.; Szijan, I.; Giliberto, F. MLPA Analysis of an Argentine Cohort of Patients with Dystrophinopathy: Association of Intron Breakpoints Hot Spots with STR Abundance in DMD Gene. J. Neurol. Sci. 2016, 365, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Sadek, A.A.; Mahmoud, S.M.; El-Aal, M.A.; Allam, A.A.; El-Halim, W.I.A. Evaluation of Cardiac Functions in Children with Duchenne Muscular Dystrophy: A Prospective Case-Control Study. Electron. Phys. 2017, 9, 5732–5739. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Hirayama, K.; Ueda, H.; Ochi, E. Two and Four Weeks of β-Hydroxy-β-Methylbutyrate (HMB) Supplementations Reduce Muscle Damage Following Eccentric Contractions. J. Am. Coll. Nutr. 2019, 38, 373–379. [Google Scholar]
- McPherron, A.C.; Lee, S.J. Double Muscling in Cattle Due to Mutations in the Myostatin Gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef]
- Shieh, P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 840–848. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of Myostatin Pathway in Neuromuscular Diseases May Explain Challenges of Anti-Myostatin Therapeutic Approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.-J. Loss of Myostatin Attenuates Severity of Muscular Dystrophy in Mdx Mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.-A.; Ahima, R.S.; Khurana, T.S. Functional Improvement of Dystrophic Muscle by Myostatin Blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Amthor, H.; Hoogaars, W.M.H. Interference with Myostatin/ActRIIB Signaling as a Therapeutic Strategy for Duchenne Muscular Dystrophy. Curr. Gene Ther. 2012, 12, 245–259. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Mura, G.; Messina, G. Therapeutic Approaches to Preserve the Musculature in Duchenne Muscular Dystrophy: The Importance of the Secondary Therapies. Exp. Cell Res. 2022, 410, 112968. [Google Scholar] [CrossRef] [PubMed]
- Mobley, C.B.; Fox, C.D.; Ferguson, B.S.; Amin, R.H.; Dalbo, V.J.; Baier, S.; Rathmacher, J.A.; Wilson, J.M.; Roberts, M.D. L-Leucine, Beta-Hydroxy-Beta-Methylbutyric Acid (HMB) and Creatine Monohydrate Prevent Myostatin-Induced Akirin-1/Mighty MRNA down-Regulation and Myotube Atrophy. J. Int. Soc. Sports Nutr. 2014, 11, 38. [Google Scholar] [CrossRef]
- Russ, D.W.; Acksel, C.; McCorkle, K.W.; Edens, N.K.; Garvey, S.M. Effects of Running Wheel Activity and Dietary HMB and B—Alanine Co-Supplementation on Muscle Quality in Aged Male Rats. J. Nutr. Health Aging 2017, 21, 554–561. [Google Scholar] [CrossRef]
- Duan, Y.H.; Zeng, L.M.; Li, F.N.; Kong, X.F.; Xu, K.; Guo, Q.P.; Wang, W.L.; Zhang, L.Y. β-Hydroxy-β-Methyl Butyrate Promotes Leucine Metabolism and Improves Muscle Fibre Composition in Growing Pigs. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Olveira, G.; Olveira, C.; Doña, E.; Palenque, F.J.; Porras, N.; Dorado, A.; Godoy, A.M.; Rubio-Martínez, E.; Rojo-Martínez, G.; Martín-Valero, R. Oral Supplement Enriched in HMB Combined with Pulmonary Rehabilitation Improves Body Composition and Health Related Quality of Life in Patients with Bronchiectasis (Prospective, Randomised Study). Clin. Nutr. Edinb. Scotl. 2016, 35, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Grant, S.C.; Lee, S.-R.; Masad, I.S.; Park, Y.-M.; Henning, P.C.; Stout, J.R.; Loenneke, J.P.; Arjmandi, B.H.; Panton, L.B.; et al. Beta-Hydroxy-Beta-Methyl-Butyrate Blunts Negative Age-Related Changes in Body Composition, Functionality and Myofiber Dimensions in Rats. J. Int. Soc. Sports Nutr. 2012, 9, 18. [Google Scholar] [CrossRef]
- Park, B.-S.; Henning, P.C.; Grant, S.C.; Lee, W.J.; Lee, S.-R.; Arjmandi, B.H.; Kim, J.-S. HMB Attenuates Muscle Loss during Sustained Energy Deficit Induced by Calorie Restriction and Endurance Exercise. Metabolism 2013, 62, 1718–1729. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E. STAT3 Signalling Is Required for Leptin Regulation of Energy Balance but Not Reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G.; Schwartz, M.W. Intracellular Signalling. Key Enzyme in Leptin-Induced Anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A.; Leinninger, G.M.; Myers, M.G. Molecular and Neural Mediators of Leptin Action. Physiol. Behav. 2008, 94, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cruz, M.; Cruz-Guzmán, O.R.; Escobar, R.E.; López-Alarcón, M. Leptin and Metabolic Syndrome in Patients with Duchenne/Becker Muscular Dystrophy. Acta Neurol. Scand. 2016, 133, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Charrout, M.; van den Bergen, J.; Koeks, Z.; Kan, H.E.; Hooijmans, M.T.; Roos, A.; Straub, V.; et al. Tracking Disease Progression Non-Invasively in Duchenne and Becker Muscular Dystrophies. J. Cachexia Sarcopenia Muscle 2018, 9, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewska, E.; Muszyński, S.; Dobrowolski, P.; Wiącek, D.; Tomczyk-Warunek, A.; Świetlicka, I.; Pierzynowski, S.G. Maternal HMB Treatment Affects Bone and Hyaline Cartilage Development in Their Weaned Piglets via the Leptin/Osteoprotegerin System. J. Anim. Physiol. Anim. Nutr. 2019, 103, 626–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Baek, M.Y.; Moon, K.Y.; Song, W.K.; Chung, C.H.; Ha, D.B.; Kang, M.S. Nitric Oxide as a Messenger Molecule for Myoblast Fusion. J. Biol. Chem. 1994, 269, 14371–14374. [Google Scholar] [CrossRef]
- Roberts, C.K.; Barnard, R.J.; Scheck, S.H.; Balon, T.W. Exercise-Stimulated Glucose Transport in Skeletal Muscle Is Nitric Oxide Dependent. Am. J. Physiol.-Endocrinol. Metab. 1997, 273, E220–E225. [Google Scholar] [CrossRef]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired Metabolic Modulation of Alpha-Adrenergic Vasoconstriction in Dystrophin-Deficient Skeletal Muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef]
- Tews, D.S. Apoptosis and Muscle Fibre Loss in Neuromuscular Disorders. Neuromuscul. Disord. 2002, 12, 613–622. [Google Scholar] [CrossRef]
- Brusa, R.; Magri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Noncoding RNAs in Duchenne and Becker Muscular Dystrophies: Role in Pathogenesis and Future Prognostic and Therapeutic Perspectives. Cell. Mol. Life Sci. 2020, 77, 4299–4313. [Google Scholar] [CrossRef]
- Kanuri, B.N.; Kanshana, J.S.; Rebello, S.C.; Pathak, P.; Gupta, A.P.; Gayen, J.R.; Jagavelu, K.; Dikshit, M. Altered Glucose and Lipid Homeostasis in Liver and Adipose Tissue Pre-Dispose Inducible NOS Knockout Mice to Insulin Resistance. Sci. Rep. 2017, 7, 41009. [Google Scholar] [CrossRef]
- Gücüyener, K.; Ergenekon, E.; Erbas, D.; Pinarli, G.; Serdaroğlu, A. The Serum Nitric Oxide Levels in Patients with Duchenne Muscular Dystrophy. Brain Dev. 2000, 22, 181–183. [Google Scholar] [CrossRef]
- Guilbaud, M.; Gentil, C.; Peccate, C.; Gargaun, E.; Holtzmann, I.; Gruszczynski, C.; Falcone, S.; Mamchaoui, K.; Ben Yaou, R.; Leturcq, F.; et al. MiR-708-5p and MiR-34c-5p Are Involved in NNOS Regulation in Dystrophic Context. Skelet. Muscle 2018, 8, 15. [Google Scholar] [CrossRef]
- Hrach, H.C.; Mangone, M. MiRNA Profiling for Early Detection and Treatment of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4638. [Google Scholar] [CrossRef]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric Oxide in Skeletal Muscle. Nature 1994, 372, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric Oxide Synthase Complexed with Dystrophin and Absent from Skeletal Muscle Sarcolemma in Duchenne Muscular Dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, R.; Ząbek, K.; Małaczewska, J.; Milewski, S.; Kaczorek-Łukowska, E. The Effects of β-Hydroxy-β-Methylbutyrate (HMB) on Chemotaxis, Phagocytosis, and Oxidative Burst of Peripheral Blood Granulocytes and Monocytes in Goats. Animals 2019, 9, 1031. [Google Scholar] [CrossRef] [PubMed]
- Szcześniak, K.A.; Ciecierska, A.; Ostaszewski, P.; Sadkowski, T. Characterisation of Equine Satellite Cell Transcriptomic Profile Response to β -Hydroxy- β -Methylbutyrate (HMB). Br. J. Nutr. 2016, 116, 1315–1325. [Google Scholar] [CrossRef]
- Yakabe, M.; Ogawa, S.; Ota, H.; Iijima, K.; Eto, M.; Ouchi, Y.; Akishita, M. Beta-Hydroxy-Beta-Methylbutyrate Inhibits Lipopolysaccharide-Induced Interleukin-6 Expression by Increasing Protein Phosphatase-1α Expression. Biomed. Sci. 2015, 1, 1–5. [Google Scholar]
- Dawra, R.K.; Sharma, O.P.; Krishna, L.; Vaid, J. The Enzymatic Profile of Urine and Plasma in Bovine Urinary Bladder Cancer (Enzootic Bovine Haematuria). Vet. Res. Commun. 1991, 15, 421–426. [Google Scholar] [CrossRef]
- Klein, R.; Nagy, O.; Tóthová, C.; Chovanová, F. Clinical and Diagnostic Significance of Lactate Dehydrogenase and Its Isoenzymes in Animals. Vet. Med. Int. 2020, 2020, 5346483. [Google Scholar] [CrossRef] [PubMed]
- Heinova, D.; Rosival, I.; Avidar, Y.; Bogin, E. Lactate Dehydrogenase Isoenzyme Distribution and Patterns in Chicken Organs. Res. Vet. Sci. 1999, 67, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Lott, N.; Nemensanszky, E.E. Lactate dehydrogenase. In Clinical Enzymology: A Case-Oriented Approach; Lott, J.A., Wolf, P.L., Sawhney, A.K., Eds.; Field and Rich/Year Book: New York, NY, USA, 1987; pp. 213–244. [Google Scholar]
- D’Amico, A.; Catteruccia, M.; Baranello, G.; Politano, L.; Govoni, A.; Previtali, S.C.; Pane, M.; D’Angelo, M.G.; Bruno, C.; Messina, S. Diagnosis of Duchenne Muscular Dystrophy in Italy in the Last Decade: Critical Issues and Areas for Improvements. Neuromuscul. Disord. 2017, 27, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, P.M.; Hubal, M.J. Exercise-Induced Muscle Damage in Humans. Am. J. Phys. Med. Rehabil. 2002, 81, S52–S69. [Google Scholar] [CrossRef]
- Bloomer, R.J. The Role of Nutritional Supplements in the Prevention and Treatment of Resistance Exercise-Induced Skeletal Muscle Injury. Sports Med. 2007, 37, 519–532. [Google Scholar] [CrossRef]
- Ostaszewski, P.; Kowalska, A.; Szarska, E.; Szpotański, P.; Cywinska, A.; Bałasińska, B.; Sadkowski, T. Effects of β-Hydroxy-β-Methylbutyrate and γ-Oryzanol on Blood Biochemical Markers in Exercising Thoroughbred Race Horses. J. Equine Vet. Sci. 2012, 32, 542–551. [Google Scholar] [CrossRef]
- He, Y.; Han, F.; Wang, J. Clinical and Genetic Variation Analysis of 97 Patients with Duchenne Muscular Dystrophy. Chin. J. Pract. Pediatr. 2019, 33–36. [Google Scholar]
- Joshi, P.R.; Sarangerel, J.; Munkhbayar, R.; Gläser, D.; Zierz, S. Exon Skipping in Duchenne Muscle Dystrophy Due to a Silent p. Ser443= Mutation in the DMD Gene. J. Clin. Neurosci. 2020, 76, 229–232. [Google Scholar] [CrossRef]
- Wang, F.; Wen, J.; Guo, B.; Wu, L.; Liu, Z.; Zaijun, Z. Behavioral, Biochemical and Pathological Characterization of a New MDX Mouse Model of Duchenne Muscular Dystrophy. J. Pharm. Biomed. Sci. 2020, 10. [Google Scholar]
- Knitter, A.E.; Panton, L.; Rathmacher, J.A.; Petersen, A.; Sharp, R. Effects of β-Hydroxy-β-Methylbutyrate on Muscle Damage after a Prolonged Run. J. Appl. Physiol. 2000, 89, 1340–1344. [Google Scholar] [CrossRef]
- Nissen, S.; Sharp, R.; Ray, M.; Rathmacher, J.A.; Rice, D.; Fuller, J.C.; Connelly, A.S.; Abumrad, N. Effect of Leucine Metabolite Beta-Hydroxy-Beta-Methylbutyrate on Muscle Metabolism during Resistance-Exercise Training. J. Appl. Physiol. 1996, 81, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- van Someren, K.A.; Edwards, A.J.; Howatson, G. Supplementation with Beta-Hydroxy-Beta-Methylbutyrate (HMB) and Alpha-Ketoisocaproic Acid (KIC) Reduces Signs and Symptoms of Exercise-Induced Muscle Damage in Man. Int. J. Sport Nutr. Exerc. Metab. 2005, 15, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, M.H.; Mohammadi, H.; Eshaghi, H.; Askari, G.; Miraghajani, M. The Effects of Beta-Hydroxy-Beta-Methylbutyrate Supplementation on Recovery Following Exercise-Induced Muscle Damage: A Systematic Review and Meta-Analysis. J. Am. Coll. Nutr. 2018, 37, 640–649. [Google Scholar] [CrossRef] [PubMed]
- de Barros, L.F.L.; Zogaib, R.; Conforti, C.A. 2 The Effect of HMB Supplementation in Professional Soccer Players. Br. J. Sports Med. 2017, 51, A2–A3. [Google Scholar]
- Osuka, Y.; Kojima, N.; Sasai, H.; Wakaba, K.; Miyauchi, D.; Tanaka, K.; Kim, H. Effects of Exercise and/or β-Hydroxy-β-Methylbutyrate Supplementation on Muscle Mass, Muscle Strength, and Physical Performance in Older Women with Low Muscle Mass: A Randomized, Double-Blind, Placebo-Controlled Trial. Am. J. Clin. Nutr. 2021, 114, 1371–1385. [Google Scholar] [CrossRef]
- Nunan, D.; Howatson, G.; van Someren, K.A. Exercise-Induced Muscle Damage Is Not Attenuated by β-Hydroxy-β-Methylbutyrate and α-Ketoisocaproic Acid Supplementation. J. Strength Cond. Res. 2010, 24, 531. [Google Scholar] [CrossRef]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological Actions of Statins: A Critical Appraisal in the Management of Cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Pleiotropic Effects of Statins: Basic Research and Clinical Perspectives. Circ. J. 2010, 74, 818–826. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. Simvastatin Offers New Prospects for the Treatment of Duchenne Muscular Dystrophy. Rare Dis. 2016, 4, e1156286. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. A New Therapeutic Effect of Simvastatin Revealed by Functional Improvement in Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 12864–12869. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.-Y.; Hanes, J. Mucus-Penetrating Nanoparticles for Drug and Gene Delivery to Mucosal Tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M. Autophagy as a New Therapeutic Target in Duchenne Muscular Dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Duplany, A.; Moncollin, V.; Mouradian, S.; Goillot, E.; Mazelin, L.; Gauthier, K.; Streichenberger, N.; Angleraux, C.; Chen, J.; et al. Lack of Muscle MTOR Kinase Activity Causes Early Onset Myopathy and Compromises Whole-Body Homeostasis. J. Cachexia Sarcopenia Muscle 2019, 10, 35–53. [Google Scholar] [CrossRef]
- Grip, O.; Janciauskiene, S.; Lindgren, S. Pravastatin Down-Regulates Inflammatory Mediators in Human Monocytes in Vitro. Eur. J. Pharmacol. 2000, 410, 83–92. [Google Scholar] [CrossRef]
- Mendes, P.; Robles, P.G.; Mathur, S. Statin-Induced Rhabdomyolysis: A Comprehensive Review of Case Reports. Physiother. Can. 2014, 66, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Saleh, Y.; Herzallah, K.; Hassanein, M.; Chang, H.T. Statin-Induced Necrotizing Autoimmune Myopathy: An Uncommon Complication of a Commonly Used Medication. J. Saudi Heart Assoc. 2019, 31, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.; Braun, L.; Rubinstein, G.; Zopp, S.; Benedix, S.; Schneider, H.; Ritzel, K.; Schilbach, K.; Schmidmaier, R.; Beuschlein, F.; et al. Patients with Low IGF-I after Curative Surgery for Cushing’s Syndrome Have an Adverse Long-Term Outcome of Hypercortisolism-Induced Myopathy. Eur. J. Endocrinol. 2021, 184, 813–821. [Google Scholar] [CrossRef]
- Kajstura, J.; Fiordaliso, F.; Andreoli, A.M.; Li, B.; Chimenti, S.; Medow, M.S.; Limana, F.; Nadal-Ginard, B.; Leri, A.; Anversa, P. IGF1 IGF1 Overexpression Inhibits the Development of Diabetic Cardiomyopathy and Angiotensin II–Mediated Oxidative Stress. Diabetes 2001, 50, 1414–1424. [Google Scholar] [CrossRef]
- Yamada, A.K.; Ferretti, R.; Matsumura, C.Y.; Antunes, L.; da Silva, C.A.; Pertille, A. Beta-Hydroxy-Beta-Methylbutyrate Associated with Low-Intensity Exercise Training Improves Skeletal Muscle Regeneration through the IGF-Akt Pathway. Braz. J. Med. Biol. Res. 2022, 55, e11597. [Google Scholar] [CrossRef]
- Laoutaris, I.D.; Adamopoulos, S.; Manginas, A.; Panagiotakos, D.B.; Cokkinos, D.V.; Dritsas, A. Inspiratory Work Capacity Is More Severely Depressed than Inspiratory Muscle Strength in Patients with Heart Failure: Novel Applications for Inspiratory Muscle Training. Int. J. Cardiol. 2016, 221, 622–626. [Google Scholar] [CrossRef]
- Rahman, F.A.; Quadrilatero, J. Mitochondrial Apoptotic Signaling Involvement in Remodeling During Myogenesis and Skeletal Muscle Atrophy. Semin. Cell Dev. Biol. 2023, 143, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Essid, S.M.; Bevington, A.; Brunskill, N.J. Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts. Int. J. Mol. Sci. 2019, 20, 1654. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, T.; Genovesi, F.; Nemmer, M.; Carling, C.; Alberti, G.; Howatson, G. Nutritional Interventions for Reducing the Signs and Symptoms of Exercise-Induced Muscle Damage and Accelerate Recovery in Athletes: Current Knowledge, Practical Application and Future Perspectives. Eur. J. Appl. Physiol. 2020, 120, 1965–1996. [Google Scholar] [CrossRef]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A Practical Guide to the Monitoring and Management of the Complications of Systemic Corticosteroid Therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice Guideline Update Summary: Corticosteroid Treatment of Duchenne Muscular Dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef]
- Florini, J.R.; Ewton, D.Z.; Magri, K.A. Hormones, Growth Factors, and Myogenic Differentiation. Annu. Rev. Physiol. 1991, 53, 201–216. [Google Scholar] [CrossRef]
- Pasquini, F.; Guerin, C.; Blake, D.; Davies, K.; Karpati, G.; Holland, P. The Effect of Glucocorticoids on the Accumulation of Utrophin by Cultured Normal and Dystrophic Human Skeletal Muscle Satellite Cells. Neuromuscul. Disord. 1995, 5, 105–114. [Google Scholar] [CrossRef]
- Anderson, J.E.; Weber, M.; Vargas, C. Deflazacort Increases Laminin Expression and Myogenic Repair, and Induces Early Persistent Functional Gain in Mdx Mouse Muscular Dystrophy. Cell Transplant. 2000, 9, 551–564. [Google Scholar] [CrossRef]
- Jacobs, S.; Bootsma, A.L.; Willems, P.W.A.; Bär, P.R.; Wokke, J.H.J. Prednisone Can Protect against Exercise-Induced Muscle Damage. J. Neurol. 1996, 243, 410–416. [Google Scholar] [CrossRef]
- Muntoni, F.; Fisher, I.; Morgan, J.E.; Abraham, D. Steroids in Duchenne Muscular Dystrophy: From Clinical Trials to Genomic Research. Neuromuscul. Disord. 2002, 12, S162–S165. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the Treatment of Duchenne Muscular Dystrophy. Cochrane Database Syst. Rev. 2016, 2016, CD003725. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Sklar, R.M.; Brown, R.H. Methylprednisolone Selectively Affects Dystrophin Expression in Human Muscle Cultures. Neurology 1993, 43, 342. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.; Petersson, S.J.; Illum, N.O.; Laugaard-Jacobsen, H.C.; Thelle, T.; Jørgensen, L.H.; Schrøder, H.D. Muscular Response to the First Three Months of Deflazacort Treatment in Boys with Duchenne Muscular Dystrophy. J. Musculoskelet. Neuronal Interact. 2017, 17, 8. [Google Scholar] [PubMed]
- Butterfield, R.J.; Kirkov, S.; Conway, K.M.; Johnson, N.; Matthews, D.; Phan, H.; Cai, B.; Paramsothy, P.; Thomas, S.; Feldkamp, M.L. Evaluation of Effects of Continued Corticosteroid Treatment on Cardiac and Pulmonary Function in Non-Ambulatory Males with Duchenne Muscular Dystrophy from MD STAR Net. Muscle Nerve 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Curtis, J.R.; Westfall, A.O.; Allison, J.; Bijlsma, J.W.; Freeman, A.; George, V.; Kovac, S.H.; Spettell, C.M.; Saag, K.G. Population-Based Assessment of Adverse Events Associated with Long-Term Glucocorticoid Use. Arthritis Care Res. 2006, 55, 420–426. [Google Scholar] [CrossRef]
- Mandel, K.; Atkinson, S.; Barr, R.D.; Pencharz, P. Skeletal Morbidity in Childhood Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2004, 22, 1215–1221. [Google Scholar] [CrossRef]
- Hochberg, Z. Mechanisms of Steroid Impairment of Growth. Horm. Res. Paediatr. 2002, 58, 33–38. [Google Scholar] [CrossRef]
- Ogawa, M.; Yoshida, N.; Satomi-Kobayashi, S.; Tsuboi, Y.; Komaki, K.; Wakida, K.; Gotake, Y.; Inoue, T.; Tanaka, H.; Yamashita, T.; et al. Efficacy of Preoperative Amino Acid Supplements on Postoperative Physical Function and Complications in Open Heart Surgery Patients: A Study Protocol for a Randomized Controlled Trial. J. Cardiol. 2019, 74, 360–365. [Google Scholar] [CrossRef]
- Kim, H.; Perlingeiro, R.C.R. Generation of Human Myogenic Progenitors from Pluripotent Stem Cells for in Vivo Regeneration. Cell. Mol. Life Sci. 2022, 79, 406. [Google Scholar] [CrossRef]
- Zheng, J.; Li, B.; Yan, Y.; Huang, X.; Zhang, E. β-Hydroxy-β-Methylbutyric Acid Promotes Repair of Sheep Myoblast Injury by Inhibiting IL-17/NF-ΚB Signaling. Int. J. Mol. Sci. 2023, 24, 444. [Google Scholar] [CrossRef] [PubMed]
- Giron, M.D.; Vílchez, J.D.; Shreeram, S.; Salto, R.; Manzano, M.; Cabrera, E.; Campos, N.; Edens, N.K.; Rueda, R.; Lopez-Pedrosa, J.M. β-Hydroxy-β-Methylbutyrate (HMB) Normalizes Dexamethasone-Induced Autophagy-Lysosomal Pathway in Skeletal Muscle. PLoS ONE 2015, 10, e0117520. [Google Scholar] [CrossRef] [PubMed]
- Landi, F.; Calvani, R.; Picca, A.; Marzetti, E. Beta-Hydroxy-Beta-Methylbutyrate and Sarcopenia: From Biological Plausibility to Clinical Evidence. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | DMD Effect | References | HMB Effect | References |
|---|---|---|---|---|
| MSTN | inhibitor | Chiu, W. et al. (2020) [81] | stimulator | Shirvani, H. et al. (2020) [82] |
| AKT1 | stimulator | Peter, A.K.; Crosbie, R.H. (2006) [47], Kornegay, J.N. et al. (2012) [83] | stimulator | Salto, R. et al. (2015) [49] Schnuck, J.K. et al. (2016) [79] |
| LEP | stimulator | Söderpalm, A.C. et al. (2007) [84] | stimulator | Świetlicka, I. et al. (2021) [85] |
| IL6 | stimulator | Rodríguez-Cruz, M. et al. (2018) [17] Gallardo, F.S. et al. (2021) [60] | inhibitor | Miyake, S. et al. (2019) [58] |
| CASP3 | positive effect | Parrotta, E.I. et al. (2020) [63] | inhibitor | Eley, H.L. et al. (2008) [10] Hao, Y. et al. (2011) [67] |
| MAPK (MAPK14) | biomarker | Cotán, D. et al. (2016) [86] | inhibitor | Eley, H.L. et al. (2008) [10] |
| MyoD family | unknown | Gonçalves, M.A.F.V. et al. (2008 and 2011) [87,88] | stimulator | Kim, J.-S. et al. (2013) [46] |
| nNOS | unknown | Tian, L.J. et al. (2014) [89] | stimulator | Peterson, A.L. et al. (1999) [90] |
| NF-κB | stimulator | Wattin, M. et al. (2018) [53] Finkel, R.S. et al. (2021) [91] | inhibitor | Smith, H.J. et al. (2005) [4] Miyake, S. et al. (2019) [58] |
| LDH | stimulator | Luce, L.N. et al. (2016) [92] Sadek, A.A. et al. (2017) [93] | unknown | Tsuchiya, Y. et al. (2019) [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorji, A.E.; Ostaszewski, P.; Urbańska, K.; Sadkowski, T. Does β-Hydroxy-β-Methylbutyrate Have Any Potential to Support the Treatment of Duchenne Muscular Dystrophy in Humans and Animals? Biomedicines 2023, 11, 2329. https://doi.org/10.3390/biomedicines11082329
Gorji AE, Ostaszewski P, Urbańska K, Sadkowski T. Does β-Hydroxy-β-Methylbutyrate Have Any Potential to Support the Treatment of Duchenne Muscular Dystrophy in Humans and Animals? Biomedicines. 2023; 11(8):2329. https://doi.org/10.3390/biomedicines11082329
Chicago/Turabian StyleGorji, Abdolvahab Ebrahimpour, Piotr Ostaszewski, Kaja Urbańska, and Tomasz Sadkowski. 2023. "Does β-Hydroxy-β-Methylbutyrate Have Any Potential to Support the Treatment of Duchenne Muscular Dystrophy in Humans and Animals?" Biomedicines 11, no. 8: 2329. https://doi.org/10.3390/biomedicines11082329
APA StyleGorji, A. E., Ostaszewski, P., Urbańska, K., & Sadkowski, T. (2023). Does β-Hydroxy-β-Methylbutyrate Have Any Potential to Support the Treatment of Duchenne Muscular Dystrophy in Humans and Animals? Biomedicines, 11(8), 2329. https://doi.org/10.3390/biomedicines11082329