
Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease


Abstract
:1. Introduction
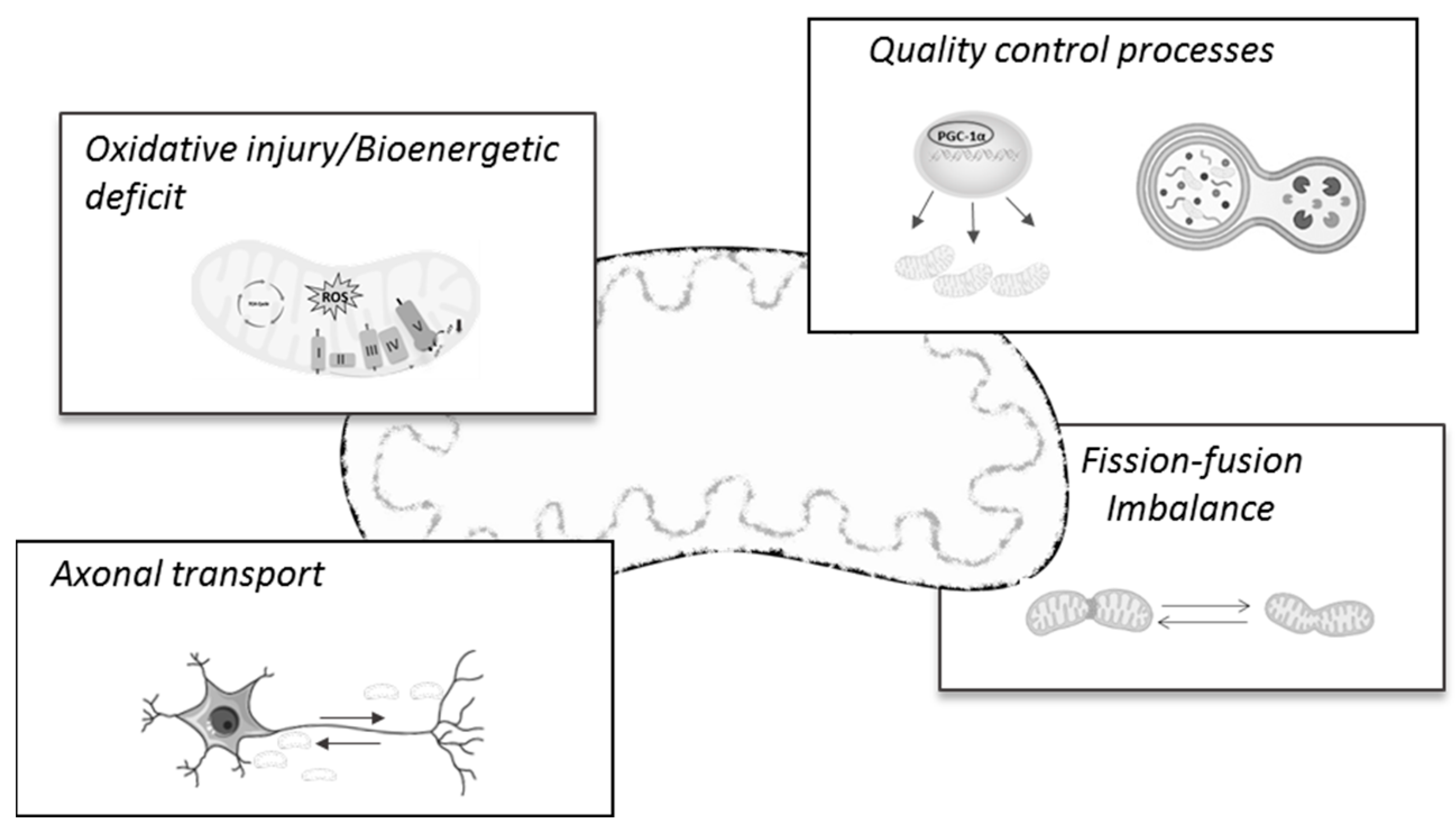
2. Mitochondria (Dys)Function in Alzheimer’s Disease: A Brief Overview of the Main Mechanisms
2.1. Relevance of Oxidative Stress and Energy (Hypo)Metabolism in AD
2.2. Mitochondrial Dynamics and Transport Alterations in AD
2.3. Mitochondrial Biogenesis and Mitophagy
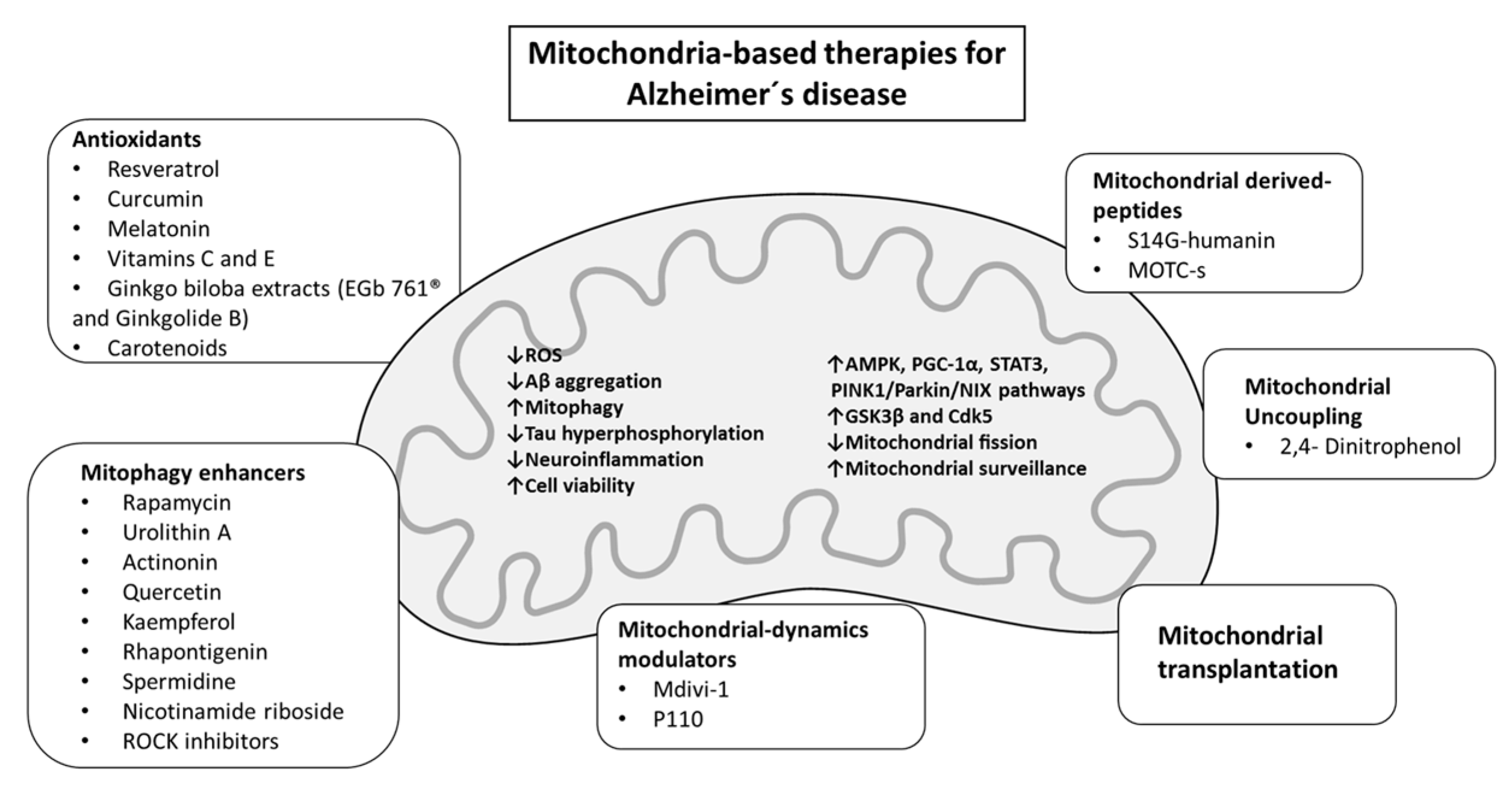
3. Mitochondria-Based Therapies for Alzheimer’s Disease
3.1. Mitochondrial Antioxidant Interventions
3.2. Mitochondrial-Derived Peptides as Therapeutic Agents
3.3. Mitochondrial Dynamics and Mitophagy-Targeting Therapy in AD
3.3.1. Mitochondria Dynamics Modulators
3.3.2. Mitophagy Enhancers
3.4. Mitochondrial Uncoupling in AD

{kind=link}
{kind=link}
| Mitochondria-Based Therapeutics | Targets and Mechanism of Action | Benefits and Limitations | References | |
|---|---|---|---|---|
| Antioxidants | Resveratrol |
| Benefits (pre-clinical):
| [142,143,144,145,146,147,148,149,150,154,155] |
| Curcumin |
| Benefits (pre-clinical):
| [158,159,160,161,162,163,164,165,166,170,171] | |
| Melatonin |
| Benefits:
| [185,190,191,192,193,194,195,196] | |
| Vitamins C and E |
| Benefits:
| [197,199] | |
| a | Carotenoids |
| Benefits (clinical):
| [200,201,202] |
| Ginkgo biloba extracts (EGb761® and Ginkgolide B) |
| Benefits (pre-clinical):
| [175,176,177,178,179,180,181,182,183] | |
| Mitochondrial-derived peptides | S14G-humanin |
| Benefits (pre-clinical):
| [206,211,214,215] |
| Colivelin |
| Benefits (pre-clinical):
| [217,218] | |
| MOTC-s |
| Benefits (pre-clinical):
| [220,221,222,223] | |
| Mitochondrial dynamics modulators | Mdivi-1 |
| Benefits (pre-clinical):
| [230,231] |
| P110 |
| Benefits (pre-clinical):
| [85,233] | |
| Mitophagy enhancers | Rapamycin |
| Benefits (pre-clinical):
| [235,236,238,239] |
| Urolithin A |
| Benefits (pre-clinical):
| [134,242,243,244,245] | |
| Actinonin |
| Benefits (pre-clinical):
| [134] | |
| Memantine |
| Benefits (pre-clinical):
| [247] | |
| Quercetin |
| Benefits (pre-clinical):
| [250,251,252,253,254] | |
| Kaempferol and rhapontigenin |
| Benefits (pre-clinical):
| [255] | |
| Spermidine |
| Benefits (pre-clinical):
| [257,260,261,262] | |
| Nicotinamide adenine dinucleotide (NAD+) percursors |
| Benefits (pre-clinical):
| [263,264,265,266] | |
| ROCK inhibitors |
| Benefits (pre-clinical):
| [274,275] | |
| Combined metabolic activators (CMA) |
| Benefits (clinical):
| [267] | |
| Mitochondrial uncoupling | Mitochondrial uncoupling proteins(UCP2, UCP4) |
| Benefits (pre-clinical):
| [279,281,282,283,284,286,287] |
| 2,4-Dinitrophenol |
| Benefits (pre-clinical):
| [280,281,282,283,284,285] | |
4. Mitochondrial Transplantation: Could the Transfer of Healthy Mitochondria Be the Solution?
5. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-D.-C.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the Path to 2025: Understanding the Alzheimer’s Disease Continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s Disease Drug-Development Pipeline: 2023. Alzheimer’s Dement. 2023, 6, 37. [Google Scholar] [CrossRef]
- Vradenburg, G. A Pivotal Moment in Alzheimer’s Disease and Dementia: How Global Unity of Purpose and Action Can Beat the Disease by 2025. Expert Rev. Neurother. 2015, 15, 73–82. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug Development in Alzheimer’s Disease: The Path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef]
- Zhang, M.; Ganz, A.B.; Rohde, S.; Rozemuller, A.J.M.; Netherlands Brain Bank; Reinders, M.J.T.; Scheltens, P.; Hulsman, M.; Hoozemans, J.J.M.; Holstege, H. Resilience and Resistance to Alzheimer’s Disease-Associated Neuropathological Substrates in Centenarians: An Age-Continuous Perspective. Neurology 2022. preprint. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The Role of Mitochondrial Function and Cellular Bioenergetics in Ageing and Disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz E Silva, O. Mitochondria, Energy, and Metabolism in Neuronal Health and Disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s Disease Is Associated with Reduced Expression of Energy Metabolism Genes in Posterior Cingulate Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial Dysfunction in Alzheimer’s Disease—A Proteomics Perspective. Expert Rev. Proteom. 2021, 18, 295–304. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial Dysfunction, Oxidative Stress, Neuroinflammation, and Metabolic Alterations in the Progression of Alzheimer’s Disease: A Meta-Analysis of in Vivo Magnetic Resonance Spectroscopy Studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Lu, Y.; Siedlak, S.L.; Fujioka, H.; Feng, H.; Perry, G.; Zhu, X. Damaged Mitochondria Coincide with Presynaptic Vesicle Loss and Abnormalities in Alzheimer’s Disease Brain. Acta Neuropathol. Commun. 2023, 11, 54. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.-B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy Crisis Precedes Global Metabolic Failure in a Novel Caenorhabditis Elegans Alzheimer Disease Model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial Dysfunction: An Early Event in Alzheimer Pathology Accumulates with Age in AD Transgenic Mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Saraiva, A.A.; Borges, M.M.; Madeira, M.D.; Tavares, M.A.; Paula-Barbosa, M.M. Mitochondrial Abnormalities in Cortical Dendrites from Patients with Alzheimer’s Disease. J. Submicrosc. Cytol. 1985, 17, 459–464. [Google Scholar]
- Paula-Barbosa, M.M.; Cardoso, R.M.; Guimaraes, M.L.; Cruz, C. Dendritic Degeneration and Regrowth in the Cerebral Cortex of Patients with Alzheimer’s Disease. J. Neurol. Sci. 1980, 45, 129–134. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, J.; Tan, M.; Albers, K.M.; Jia, J. Mitochondrial Fission Proteins in Peripheral Blood Lymphocytes Are Potential Biomarkers for Alzheimer’s Disease. Eur. J. Neurol. 2012, 19, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ponce, D.P.; Osorio-Fuentealba, C.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front. Neurosci. 2017, 11, 553. [Google Scholar] [CrossRef]
- Mahapatra, G.; Gao, Z.; Bateman, J.R.; Lockhart, S.N.; Bergstrom, J.; DeWitt, A.R.; Piloso, J.E.; Kramer, P.A.; Gonzalez-Armenta, J.L.; Amick, K.A.; et al. Blood-Based Bioenergetic Profiling Reveals Differences in Mitochondrial Function Associated with Cognitive Performance and Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral Levels of Glutathione and Protein Oxidation as Markers in the Development of Alzheimer’s Disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef]
- Padurariu, M.; Ciobica, A.; Hritcu, L.; Stoica, B.; Bild, W.; Stefanescu, C. Changes of Some Oxidative Stress Markers in the Serum of Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Neurosci. Lett. 2010, 469, 6–10. [Google Scholar] [CrossRef]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; Loureiro, A.P.d.M.; de Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral Oxidative Stress Biomarkers in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef]
- López, N.; Tormo, C.; De Blas, I.; Llinares, I.; Alom, J. Oxidative Stress in Alzheimer’s Disease and Mild Cognitive Impairment with High Sensitivity and Specificity. J. Alzheimer’s Dis. 2013, 33, 823–829. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox Metabolism: ROS as Specific Molecular Regulators of Cell Signaling and Function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M. Brain Regional Correspondence between Alzheimer’s Disease Histopathology and Biomarkers of Protein Oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M., Jr.; Turner, D.M.; Markesbery, W.R.; Butterfield, A.D. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell. Mol. Med. 2009, 13, 2019–2029. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-Hydroxynonenal, a Product of Lipid Peroxidation, Is Increased in the Brain in Alzheimer’s Disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the Lipid Peroxidation Product, HNE, in the Pathogenesis and Progression of Alzheimer’s Disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased Peroxidation and Reduced Antioxidant Enzyme Activity in Alzheimer’s Disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Fang, D.; Zhang, Z.; Li, H.; Yu, Q.; Douglas, J.T.; Bratasz, A.; Kuppusamy, P.; Yan, S.S. Increased Electron Paramagnetic Resonance Signal Correlates with Mitochondrial Dysfunction and Oxidative Stress in an Alzheimer’s Disease Mouse Brain. J. Alzheimer’s Dis. 2016, 51, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W.; Tabaton, M.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. The Earliest Stage of Cognitive Impairment in Transition from Normal Aging to Alzheimer Disease Is Marked by Prominent RNA Oxidation in Vulnerable Neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidatively Modified RNA in Mild Cognitive Impairment. Neurobiol. Dis. 2008, 29, 169–175. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-Derived Reactive Oxygen Species Lead to Enhanced Amyloid Beta Formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef]
- Wilkins, H.M. Interactions between Amyloid, Amyloid Precursor Protein, and Mitochondria. Biochem. Soc. Trans. 2023, 51, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial Amyloid-Beta Levels Are Associated with the Extent of Mitochondrial Dysfunction in Different Brain Regions and the Degree of Cognitive Impairment in Alzheimer’s Transgenic Mice. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S535–S550. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The Amyloid Beta-Peptide Is Imported into Mitochondria via the TOM Import Machinery and Localized to Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. J. Alzheimer’s Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.-C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD Enhances Abeta-Induced Cell Stress via Mitochondrial Dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar] [CrossRef]
- Fang, D.; Wang, Y.; Zhang, Z.; Du, H.; Yan, S.; Sun, Q.; Zhong, C.; Wu, L.; Vangavaragu, J.R.; Yan, S.; et al. Increased Neuronal PreP Activity Reduces Aβ Accumulation, Attenuates Neuroinflammation and Improves Mitochondrial and Synaptic Function in Alzheimer Disease’s Mouse Model. Hum. Mol. Genet. 2015, 24, 5198–5210. [Google Scholar] [CrossRef]
- Dou, Y.; Tan, Y. Presequence Protease Reverses Mitochondria-Specific Amyloid-β-Induced Mitophagy to Protect Mitochondria. FASEB J. 2023, 37, e22890. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Troutwine, B.R.; Menta, B.W.; Manley, S.J.; Strope, T.A.; Lysaker, C.R.; Swerdlow, R.H. Mitochondrial Membrane Potential Influences Amyloid-β Protein Precursor Localization and Amyloid-β Secretion. J. Alzheimer’s Dis. 2022, 85, 381–394. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging. 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain. 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between Mitochondrial Reactive Oxygen Species and Cellular Glucose Metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid Beta-Peptide Impairs Glucose Transport in Hippocampal and Cortical Neurons: Involvement of Membrane Lipid Peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of Nitrated Proteins in Alzheimer’s Disease Brain Using a Redox Proteomics Approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic Identification of HNE-Bound Proteins in Early Alzheimer Disease: Insights into the Role of Lipid Peroxidation in the Progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox Proteomics Identification of 4-Hydroxynonenal-Modified Brain Proteins in Alzheimer’s Disease: Role of Lipid Peroxidation in Alzheimer’s Disease Pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Redox Proteomics and Amyloid β-Peptide: Insights into Alzheimer Disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET Changes in Brain Glucose Metabolism from Normal Cognition to Pathologically Verified Alzheimer’s Disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal Hypometabolism Predicts Cognitive Decline from Normal Aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Brain Glucose Metabolism in the Early and Specific Diagnosis of Alzheimer’s Disease. FDG-PET Studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional Brain Abnormalities in Young Adults at Genetic Risk for Late-Onset Alzheimer’s Dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for Brain Glucose Dysregulation in Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In Vivo Mitochondrial and Glycolytic Impairments in Patients with Alzheimer Disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-delCastillo, M.; Sun, R.C.; et al. Hippocampal Disruptions of Synaptic and Astrocyte Metabolism Are Primary Events of Early Amyloid Pathology in the 5xFAD Mouse Model of Alzheimer’s Disease. Cell Death Dis. 2021, 12, 954. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation Status of Pyruvate Dehydrogenase Distinguishes Metabolic Phenotypes of Cultured Rat Brain Astrocytes and Neurons. Glia 2010, 58, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Laughton, J.D.; Bittar, P.; Charnay, Y.; Pellerin, L.; Kovari, E.; Magistretti, P.J.; Bouras, C. Metabolic Compartmentalization in the Human Cortex and Hippocampus: Evidence for a Cell- and Region-Specific Localization of Lactate Dehydrogenase 5 and Pyruvate Dehydrogenase. BMC Neurosci. 2007, 8, 35. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.-K.; Merle, M.; Magistretti, P.J.; Pellerin, L. Feeding Active Neurons: (Re)Emergence of a Nursing Role for Astrocytes. J. Physiol. Paris 2002, 96, 273–282. [Google Scholar] [CrossRef]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Gutierrez, C.; Bonora, N.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Bates, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Abrogating Mitochondrial ROS in Neurons or Astrocytes Reveals Cell-Specific Impact on Mouse Behaviour. Redox Biol. 2021, 41, 101917. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.-A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.-S.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Blazey, T.; Metcalf, N.V.; McAvoy, M.P.; Strain, J.F.; Rahmani, M.; Durbin, T.J.; Xiong, C.; Benzinger, T.L.-S.; Morris, J.C.; et al. Brain Aerobic Glycolysis and Resilience in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2212256120. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic Glycolysis and Tau Deposition in Preclinical Alzheimer’s Disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal Interaction between the Mitochondrial Fission Protein Drp1 and Hyperphosphorylated Tau in Alzheimer’s Disease Neurons: Implications for Mitochondrial Dysfunction and Neuronal Damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 Interaction Mediates Mitochondrial Dysfunction, Bioenergetic Failure and Cognitive Decline in Alzheimer’s Disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-Beta Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates Beta-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Espino de la Fuente-Muñoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermúdez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. [Google Scholar] [CrossRef]
- Guo, M.-Y.; Shang, L.; Hu, Y.-Y.; Jiang, L.-P.; Wan, Y.-Y.; Zhou, Q.-Q.; Zhang, K.; Liao, H.-F.; Yi, J.-L.; Han, X.-J. The Role of Cdk5-Mediated Drp1 Phosphorylation in Aβ1-42 Induced Mitochondrial Fission and Neuronal Apoptosis. J. Cell. Biochem. 2018, 119, 4815–4825. [Google Scholar] [CrossRef]
- Xu, D.; Yang, P.; Yang, Z.-J.; Li, Q.-G.; Ouyang, Y.-T.; Yu, T.; Shangguan, J.-H.; Wan, Y.-Y.; Jiang, L.-P.; Qu, X.-H.; et al. Blockage of Drp1 Phosphorylation at Ser579 Protects Neurons against Aβ1-42-induced Degeneration. Mol. Med. Rep. 2021, 24, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered Brain Energetics Induces Mitochondrial Fission Arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [PubMed]
- Tyumentsev, M.A.; Stefanova, N.A.; Kiseleva, E.V.; Kolosova, N.G. Mitochondria with Morphology Characteristic for Alzheimer’s Disease Patients Are Found in the Brain of OXYS Rats. Biochem. Biokhimiia 2018, 83, 1083–1088. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F.T. Ultrastructural Evidence for Impaired Mitochondrial Fission in the Aged Rhesus Monkey Dorsolateral Prefrontal Cortex. Neurobiol. Aging 2017, 51, 9–18. [Google Scholar] [CrossRef]
- Panes, J.; Nguyen, T.K.O.; Gao, H.; Christensen, T.A.; Stojakovic, A.; Trushin, S.; Salisbury, J.L.; Fuentealba, J.; Trushina, E. Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells 2023, 12, 1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer’s Disease Patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; García, E.; Perry, G.; Avila, J.; García-Escudero, V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef]
- Drabik, K.; Piecyk, K.; Wolny, A.; Szulc-Dąbrowska, L.; Dębska-Vielhaber, G.; Vielhaber, S.; Duszyński, J.; Malińska, D.; Szczepanowska, J. Adaptation of Mitochondrial Network Dynamics and Velocity of Mitochondrial Movement to Chronic Stress Present in Fibroblasts Derived from Patients with Sporadic Form of Alzheimer’s Disease. FASEB J. 2021, 35, e21586. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial Dynamics and Transport in Alzheimer’s Disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Vevea, J.D.; Chapman, E.R. Mitofusin 2 Sustains the Axonal Mitochondrial Network to Support Presynaptic Ca2+ Homeostasis and the Synaptic Vesicle Cycle in Rat Hippocampal Axons. J. Neurosci. 2023, 43, 3421–3438. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Sheng, Z.-H. Regulation of Mitochondrial Transport in Neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Amyloid Beta Impairs Mitochondrial Anterograde Transport and Degenerates Synapses in Alzheimer’s Disease Neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired Mitochondrial Biogenesis, Defective Axonal Transport of Mitochondria, Abnormal Mitochondrial Dynamics and Synaptic Degeneration in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Seo, N.-Y.; Kim, G.H.; Noh, J.E.; Shin, J.W.; Lee, C.H.; Lee, K.J. Selective Regional Loss of Cortical Synapses Lacking Presynaptic Mitochondria in the 5xFAD Mouse Model. Front. Neuroanat. 2021, 15, 690168. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.-A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-Specific Depletion of Synaptic Mitochondria in the Brains of Patients with Alzheimer’s Disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Dolan, P.J.; Jin, Y.N.; Johnson, G.V.W. Truncated Tau and Aβ Cooperatively Impair Mitochondria in Primary Neurons. Neurobiol. Aging 2012, 33, e25–e35. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.-Q.; Mucke, L. Tau Reduction Prevents Aβ-Induced Axonal Transport Deficits by Blocking Activation of GSK3β. J. Cell Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen Synthase Kinase 3 Phosphorylates Kinesin Light Chains and Negatively Regulates Kinesin-Based Motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef]
- Sabui, A.; Biswas, M.; Somvanshi, P.R.; Kandagiri, P.; Gorla, M.; Mohammed, F.; Tammineni, P. Decreased Anterograde Transport Coupled with Sustained Retrograde Transport Contributes to Reduced Axonal Mitochondrial Density in Tauopathy Neurons. Front. Mol. Neurosci. 2022, 15, 927195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tian, J.; Chen, H.; Du, H.; Guo, L. Amyloid Beta-Mediated KIF5A Deficiency Disrupts Anterograde Axonal Mitochondrial Movement. Neurobiol. Dis. 2019, 127, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Iijima-Ando, K.; Sekiya, M.; Maruko-Otake, A.; Ohtake, Y.; Suzuki, E.; Lu, B.; Iijima, K.M. Loss of Axonal Mitochondria Promotes Tau-Mediated Neurodegeneration and Alzheimer’s Disease-Related Tau Phosphorylation via PAR-1. PLoS Genet. 2012, 8, e1002918. [Google Scholar] [CrossRef] [PubMed]
- Panchal, K.; Tiwari, A.K. Miro, a Rho GTPase Genetically Interacts with Alzheimer’s Disease-Associated Genes (Tau, Aβ42 and Appl) in Drosophila Melanogaster. Biol. Open 2020, 9, bio049569. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial Biogenesis and Clearance: A Balancing Act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.-D. Mitochondrial Biogenesis: An Update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional Activators and Coactivators in the Nuclear Control of Mitochondrial Function in Mammalian Cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial Biogenesis in Neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Singulani, M.P.; Pereira, C.P.M.; Ferreira, A.F.F.; Garcia, P.C.; Ferrari, G.D.; Alberici, L.C.; Britto, L.R. Impairment of PGC-1α-Mediated Mitochondrial Biogenesis Precedes Mitochondrial Dysfunction and Alzheimer’s Pathology in the 3xTg Mouse Model of Alzheimer’s Disease. Exp. Gerontol. 2020, 133, 110882. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Pedrós, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallàs, M.; Vázquez-Carrera, M.; Casadesús, G.; et al. Early Alterations in Energy Metabolism in the Hippocampus of APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.F.; Genebra, T.; Rego, A.C.; Rodrigues, C.M.P.; Solá, S. Amyloid β Peptide Compromises Neural Stem Cell Fate by Irreversibly Disturbing Mitochondrial Oxidative State and Blocking Mitochondrial Biogenesis and Dynamics. Mol. Neurobiol. 2019, 56, 3922–3936. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-Beta-Induced Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Dong, W.; Wang, F.; Guo, W.; Zheng, X.; Chen, Y.; Zhang, W.; Shi, H. Aβ25-35 Suppresses Mitochondrial Biogenesis in Primary Hippocampal Neurons. Cell. Mol. Neurobiol. 2016, 36, 83–91. [Google Scholar] [CrossRef]
- Jia, J.; Yin, J.; Zhang, Y.; Xu, G.; Wang, M.; Jiang, H.; Li, L.; Zeng, X.; Zhu, D. Thioredoxin-1 Promotes Mitochondrial Biogenesis Through Regulating AMPK/Sirt1/PGC1α Pathway in Alzheimer’s Disease. ASN Neuro 2023, 15, 17590914231159226. [Google Scholar] [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial Clearance: Mechanisms and Roles in Cellular Fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin Is Activated by PINK1-Dependent Phosphorylation of Ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 Enhancement Is Able to Compensate Mitophagy Alterations Found in Sporadic Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-Associated Tau Impairs Mitophagy by Inhibiting Parkin Translocation to Mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal Mutant APP and Amyloid Beta-Induced Cognitive Decline, Dendritic Spine Loss, Defective Autophagy, Mitophagy and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.-C.; Wang, Z.; Luo, Y.; Zhang, X.; Liu, X.-P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau Accumulation Impairs Mitophagy via Increasing Mitochondrial Membrane Potential and Reducing Mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and Amyloid Beta-Induced Defective Autophagy, Mitophagy, Mitochondrial Structural and Functional Changes and Synaptic Damage in Hippocampal Neurons from Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of Amyloid Precursor Protein C-Terminal Fragments Triggers Mitochondrial Structure, Function, and Mitophagy Defects in Alzheimer’s Disease Models and Human Brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Drygalski, K.; Fereniec, E.; Koryciński, K.; Chomentowski, A.; Kiełczewska, A.; Odrzygóźdź, C.; Modzelewska, B. Resveratrol and Alzheimer’s Disease. From Molecular Pathophysiology to Clinical Trials. Exp. Gerontol. 2018, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Rivière, C.; Richard, T.; Vitrac, X.; Mérillon, J.-M.; Valls, J.; Monti, J.-P. New Polyphenols Active on Beta-Amyloid Aggregation. Bioorg. Med. Chem. Lett. 2008, 18, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol Attenuates Oxidative Damage through Activating Mitophagy in an in Vitro Model of Alzheimer’s Disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef]
- Han, Y.-S.; Zheng, W.-H.; Bastianetto, S.; Chabot, J.-G.; Quirion, R. Neuroprotective Effects of Resveratrol against Beta-Amyloid-Induced Neurotoxicity in Rat Hippocampal Neurons: Involvement of Protein Kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.-C.; Nicol, C.J.; Cheng, Y.-C. Resveratrol Activation of AMPK-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Inflammation and Oxidative Stress. Neurochem. Int. 2018, 115, 1–10. [Google Scholar] [CrossRef]
- Abozaid, O.A.R.; Sallam, M.W.; El-Sonbaty, S.; Aziza, S.; Emad, B.; Ahmed, E.S.A. Resveratrol-Selenium Nanoparticles Alleviate Neuroinflammation and Neurotoxicity in a Rat Model of Alzheimer’s Disease by Regulating Sirt1/MiRNA-134/GSK3β Expression. Biol. Trace Elem. Res. 2022, 200, 5104–5114. [Google Scholar] [CrossRef]
- Savaskan, E.; Olivieri, G.; Meier, F.; Seifritz, E.; Wirz-Justice, A.; Müller-Spahn, F. Red Wine Ingredient Resveratrol Protects from Beta-Amyloid Neurotoxicity. Gerontology 2003, 49, 380–383. [Google Scholar] [CrossRef]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol Mitigates Lipopolysaccharide- and Aβ-Mediated Microglial Inflammation by Inhibiting the TLR4/NF-ΚB/STAT Signaling Cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab. Dispos. Biol. Fate Chem. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Walia, V.; Kaushik, D.; Mittal, V.; Kumar, K.; Verma, R.; Parashar, J.; Akter, R.; Rahman, M.H.; Bhatia, S.; Al-Harrasi, A.; et al. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Mol. Neurobiol. 2022, 59, 657–680. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Lei, H.; Zhang, D. Recent Progress in Nanotechnology-Based Drug Carriers for Resveratrol Delivery. Drug Deliv. 2023, 30, 2174206. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chu, X.; Cui, L.; Fu, S.; Gao, C.; Li, Y.; Sun, B. Neuronal Mitochondria-Targeted Therapy for Alzheimer’s Disease by Systemic Delivery of Resveratrol Using Dual-Modified Novel Biomimetic Nanosystems. Drug Deliv. 2020, 27, 502–518. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, N.; Desquiret-Dumas, V.; Leman, G.; Chupin, S.; Baron, S.; Nivet-Antoine, V.; Vessières, E.; Ayer, A.; Henrion, D.; Lenaers, G.; et al. Resveratrol Directly Binds to Mitochondrial Complex I and Increases Oxidative Stress in Brain Mitochondria of Aged Mice. PLoS ONE 2015, 10, e0144290. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Graier, W.F. Dosis Facit Sanitatem-Concentration-Dependent Effects of Resveratrol on Mitochondria. Nutrients 2017, 9, 1117. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Zinjarde, S.; Bhargava, S.; Rajamohanan, P.R.; Ravikumar, A. Discovering Bisdemethoxycurcumin from Curcuma Longa Rhizome as a Potent Small Molecule Inhibitor of Human Pancreatic α-Amylase, a Target for Type-2 Diabetes. Food Chem. 2012, 135, 2638–2642. [Google Scholar] [CrossRef]
- Sivani, B.M.; Azzeh, M.; Patnaik, R.; Pantea Stoian, A.; Rizzo, M.; Banerjee, Y. Reconnoitering the Therapeutic Role of Curcumin in Disease Prevention and Treatment: Lessons Learnt and Future Directions. Metabolites 2022, 12, 639. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s Beta-Amyloid Fibrils in Vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids Enhance Amyloid-Beta Uptake by Macrophages of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2006, 10, 1–7. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Epigallocatechin-3-Gallate and Curcumin Suppress Amyloid Beta-Induced Beta-Site APP Cleaving Enzyme-1 Upregulation. Neuroreport 2008, 19, 1329–1333. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin Labels Amyloid Pathology in Vivo, Disrupts Existing Plaques, and Partially Restores Distorted Neurites in an Alzheimer Mouse Model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in Vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- ELBini-Dhouib, I.; Doghri, R.; Ellefi, A.; Degrach, I.; Srairi-Abid, N.; Gati, A. Curcumin Attenuated Neurotoxicity in Sporadic Animal Model of Alzheimer’s Disease. Molecules 2021, 26, 3011. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Kandimalla, R.; Kuruva, C.S. Protective Effects of a Natural Product, Curcumin, against Amyloid β Induced Mitochondrial and Synaptic Toxicities in Alzheimer’s Disease. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2016, 64, 1220–1234. [Google Scholar] [CrossRef]
- Song, X.-J.; Zhou, H.-Y.; Sun, Y.-X.; Huang, H.-C. Inhibitory Effects of Curcumin on H2O2-Induced Cell Damage and APP Expression and Processing in SH-SY5Y Cells Transfected with APP Gene with Swedish Mutation. Mol. Biol. Rep. 2020, 47, 2047–2059. [Google Scholar] [CrossRef]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients with Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-Week Randomized, Double Blind, Placebo-Controlled Study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Huang, H.-C.; Xu, K.; Jiang, Z.-F. Curcumin-Mediated Neuroprotection against Amyloid-β-Induced Mitochondrial Dysfunction Involves the Inhibition of GSK-3β. J. Alzheimer’s Dis. 2012, 32, 981–996. [Google Scholar] [CrossRef]
- Cox, K.H.M.; Pipingas, A.; Scholey, A.B. Investigation of the Effects of Solid Lipid Curcumin on Cognition and Mood in a Healthy Older Population. J. Psychopharmacol. Oxf. Engl. 2015, 29, 642–651. [Google Scholar] [CrossRef]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.-P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Y.; Sun, J.; Han, Y.; Gong, W.; Li, Y.; Feng, Y.; Wang, H.; Yang, M.; Li, Z.; et al. Neuronal Mitochondria-Targeted Delivery of Curcumin by Biomimetic Engineered Nanosystems in Alzheimer’s Disease Mice. Acta Biomater. 2020, 108, 285–299. [Google Scholar] [CrossRef]
- Kandiah, N.; Ong, P.A.; Yuda, T.; Ng, L.-L.; Mamun, K.; Merchant, R.A.; Chen, C.; Dominguez, J.; Marasigan, S.; Ampil, E.; et al. Treatment of dementia and mild cognitive impairment with or without cerebrovascular disease: Expert consensus on the use of Ginkgo biloba extract, EGb 761®. CNS Neurosci. Ther. 2019, 25, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.; Eckert, A.; Eckert, G.P.; Fink, H.; Friedland, K.; Gauthier, S.; Hoerr, R.; Ihl, R.; Kasper, S.; Möller, H.-J. Therapeutic efficacy of the Ginkgo special extract EGb761® within the framework of the mitochondrial cascade hypothesis of Alzheimer’s disease. World J. Biol. Psychiatry 2019, 20, 173–189. [Google Scholar] [CrossRef]
- Tchantchou, F.; Xu, Y.; Wu, Y.; Christen, Y.; Luo, Y. EGb 761 enhances adult hippocampal neurogenesis and phosphorylation of CREB in transgenic mouse model of Alzheimer’s disease. FASEB J. 2007, 21, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Zhang, C.; Danielsen, M.; Li, Q.; Chen, W.; Chan, Y.; Li, Y. EGb761 improves cognitive function and regulates inflammatory responses in the APP/PS1 mouse. Exp. Gerontol. 2016, 81, 92–100. [Google Scholar] [CrossRef]
- Colciaghi, F.; Borroni, B.; Zimmermann, M.; Bellone, C.; Longhi, A.; Padovani, A.; Cattabeni, F.; Christen, Y.; Di Luca, M. Amyloid precursor protein metabolism is regulated toward alpha-secretase pathway by Ginkgo biloba extracts. Neurobiol. Dis. 2004, 16, 454–460. [Google Scholar] [CrossRef]
- Ge, W.; Ren, C.; Xing, L.; Guan, L.; Zhang, C.; Sun, X.; Wang, G.; Niu, H.; Qun, S. Ginkgo biloba extract improves cognitive function and increases neurogenesis by reducing Aβ pathology in 5×FAD mice. Am. J. Transl. Res. 2021, 13, 1471–1482. [Google Scholar]
- Morató, X.; Marquié, M.; Tartari, J.P.; Lafuente, A.; Abdelnour, C.; Alegret, M.; Jofresa, S.; Buendía, M.; Pancho, A.; Aguilera, N.; et al. A randomized, open-label clinical trial in mild cognitive impairment with EGb 761 examining blood markers of inflammation and oxidative stress. Sci. Rep. 2023, 13, 5406. [Google Scholar] [CrossRef]
- Gill, I.; Kaur, S.; Kaur, N.; Dhiman, M.; Mantha, A.K. Phytochemical Ginkgolide B Attenuates Amyloid-β1-42 Induced Oxidative Damage and Altered Cellular Responses in Human Neuroblastoma SH-SY5Y Cells. J. Alzheimer’s Dis. 2017, 60 (Suppl. 1), S25–S40. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B inactivates the NLRP3 inflammasome by promoting autophagic degradation to improve learning and memory impairment in Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 329–341. [Google Scholar] [CrossRef]
- Liu, J.; Ye, T.; Zhang, Y.; Zhang, R.; Kong, Y.; Zhang, Y.; Sun, J. Protective Effect of Ginkgolide B against Cognitive Impairment in Mice via Regulation of Gut Microbiota. J. Agric. Food Chem. 2021, 69, 12230–12240. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B protects against cognitive impairment in senescence-accelerated P8 mice by mitigating oxidative stress, inflammation and ferroptosis. Biochem. Biophys. Res. Commun. 2021, 572, 7–14. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central Organelles for Melatonin’s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [PubMed]
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.-M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211. [Google Scholar] [CrossRef]
- Maurizi, C.P. Loss of Intraventricular Fluid Melatonin Can Explain the Neuropathology of Alzheimer’s Disease. Med. Hypotheses 1997, 49, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Liu, R.-Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early Neuropathological Alzheimer’s Changes in Aged Individuals Are Accompanied by Decreased Cerebrospinal Fluid Melatonin Levels. J. Pineal. Res. 2003, 35, 125–130. [Google Scholar] [CrossRef]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.; Qu, Z.; Qin, C.; Zhang, J. Melatonin Alleviates Behavioral Deficits Associated with Apoptosis and Cholinergic System Dysfunction in the APP 695 Transgenic Mouse Model of Alzheimer’s Disease. J. Pineal. Res. 2004, 37, 129–136. [Google Scholar] [CrossRef]
- Olcese, J.M.; Cao, C.; Mori, T.; Mamcarz, M.B.; Maxwell, A.; Runfeldt, M.J.; Wang, L.; Zhang, C.; Lin, X.; Zhang, G.; et al. Protection against Cognitive Deficits and Markers of Neurodegeneration by Long-Term Oral Administration of Melatonin in a Transgenic Model of Alzheimer Disease. J. Pineal. Res. 2009, 47, 82–96. [Google Scholar] [CrossRef]
- Cheng, Y.; Feng, Z.; Zhang, Q.; Zhang, J. Beneficial Effects of Melatonin in Experimental Models of Alzheimer Disease. Acta Pharmacol. Sin. 2006, 27, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J. Early Melatonin Supplementation Alleviates Oxidative Stress in a Transgenic Mouse Model of Alzheimer’s Disease. Free Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef]
- Chen, C.; Yang, C.; Wang, J.; Huang, X.; Yu, H.; Li, S.; Li, S.; Zhang, Z.; Liu, J.; Yang, X.; et al. Melatonin Ameliorates Cognitive Deficits through Improving Mitophagy in a Mouse Model of Alzheimer’s Disease. J. Pineal. Res. 2021, 71, e12774. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.-L.; Dammer, E.B.; Li, C.-B.; Xu, G.; Chen, S.-D.; Wang, G. Melatonin for Sleep Disorders and Cognition in Dementia: A Meta-Analysis of Randomized Controlled Trials. Am. J. Alzheimer’s Dis. Other Demen. 2015, 30, 439–447. [Google Scholar] [CrossRef]
- Sumsuzzman, D.M.; Choi, J.; Jin, Y.; Hong, Y. Neurocognitive Effects of Melatonin Treatment in Healthy Adults and Individuals with Alzheimer’s Disease and Insomnia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Neurosci. Biobehav. Rev. 2021, 127, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguilar, M.A.; Ramírez-Salado, I.; Guevara, M.A.; Hernández-González, M.; Benitez-King, G. Melatonin Effects on EEG Activity During Sleep Onset in Mild-to-Moderate Alzheimer’s Disease: A Pilot Study. J. Alzheimer’s Dis. Rep. 2018, 2, 55–65. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on Prolonged-Release Melatonin for Cognitive Function and Sleep in Mild to Moderate Alzheimer’s Disease: A 6-Month, Randomized, Placebo-Controlled, Multicenter Trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Nelson, V.A.; Davila, H.; Ratner, E.; Fink, H.A.; Hemmy, L.S.; McCarten, J.R.; Barclay, T.R.; Brasure, M.; Kane, R.L. Over-the-Counter Supplement Interventions to Prevent Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer-Type Dementia: A Systematic Review. Ann. Intern. Med. 2018, 168, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Manochkumar, J.; Doss, C.G.P.; El-Seedi, H.R.; Efferth, T.; Ramamoorthy, S. The neuroprotective potential of carotenoids in vitro and in vivo. Phytomedicine 2021, 91, 153676. [Google Scholar] [CrossRef]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s Disease: What Do We Know so Far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef]
- Lu, Y.; An, Y.; Guo, J.; Zhang, X.; Wang, H.; Rong, H.; Xiao, R. Dietary Intake of Nutrients and Lifestyle Affect the Risk of Mild Cognitive Impairment in the Chinese Elderly Population: A Cross-Sectional Study. Front. Behav. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef]
- Kesse-Guyot, E.; Andreeva, V.A.; Ducros, V.; Jeandel, C.; Julia, C.; Hercberg, S.; Galan, P. Carotenoid-rich dietary patterns during midlife and subsequent cognitive function. Br. J. Nutr. 2014, 111, 915–923. [Google Scholar] [CrossRef]
- Yuan, C.; Chen, H.; Wang, Y.; Schneider, J.A.; Willett, W.C.; Morris, M.C. Dietary carotenoids related to risk of incident Alzheimer dementia (AD) and brain AD neuropathology: A community-based cohort of older adults. Am. J. Clin. Nutr. 2021, 113, 200–208. [Google Scholar] [CrossRef]
- Nolan, J.M.; Power, R.; Mulcahy, R. Supplementation With Carotenoids, Omega-3 Fatty Acids, and Vitamin E Has a Positive Effect on the Symptoms and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 90, 233–249. [Google Scholar] [CrossRef]
- Miller, B.; Kim, S.-J.; Kumagai, H.; Yen, K.; Cohen, P. Mitochondria-Derived Peptides in Aging and Healthspan. J. Clin. Investig. 2022, 132, e158449. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A Rescue Factor Abolishing Neuronal Cell Death by a Wide Spectrum of Familial Alzheimer’s Disease Genes and Abeta. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef]
- Yen, K.; Wan, J.; Mehta, H.H.; Miller, B.; Christensen, A.; Levine, M.E.; Salomon, M.P.; Brandhorst, S.; Xiao, J.; Kim, S.-J.; et al. Humanin Prevents Age-Related Cognitive Decline in Mice and Is Associated with Improved Cognitive Age in Humans. Sci. Rep. 2018, 8, 14212. [Google Scholar] [CrossRef]
- Tajima, H.; Kawasumi, M.; Chiba, T.; Yamada, M.; Yamashita, K.; Nawa, M.; Kita, Y.; Kouyama, K.; Aiso, S.; Matsuoka, M.; et al. A Humanin Derivative, S14G-HN, Prevents Amyloid-Beta-Induced Memory Impairment in Mice. J. Neurosci. Res. 2005, 79, 714–723. [Google Scholar] [CrossRef]
- Niikura, T.; Sidahmed, E.; Hirata-Fukae, C.; Aisen, P.S.; Matsuoka, Y. A Humanin Derivative Reduces Amyloid Beta Accumulation and Ameliorates Memory Deficit in Triple Transgenic Mice. PLoS ONE 2011, 6, e16259. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, W.; Li, Z.; Hao, J.; Zhang, Z.; Liu, L.; Mao, N.; Miao, J.; Zhang, L. S14G-Humanin Improves Cognitive Deficits and Reduces Amyloid Pathology in the Middle-Aged APPswe/PS1dE9 Mice. Pharmacol. Biochem. Behav. 2012, 100, 361–369. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin Inhibits Neuronal Cell Death by Interacting with a Cytokine Receptor Complex or Complexes Involving CNTF Receptor Alpha/WSX-1/Gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef]
- Kim, S.-J.; Guerrero, N.; Wassef, G.; Xiao, J.; Mehta, H.H.; Cohen, P.; Yen, K. The Mitochondrial-Derived Peptide Humanin Activates the ERK1/2, AKT, and STAT3 Signaling Pathways and Has Age-Dependent Signaling Differences in the Hippocampus. Oncotarget 2016, 7, 46899–46912. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, T.; Wang, W.-X.; Xu, J.-H.; Yang, P.-B.; Lu, H.-X.; Sun, Q.-R.; Hu, H.-T. Protective Effects of [Gly14]-Humanin on Beta-Amyloid-Induced PC12 Cell Death by Preventing Mitochondrial Dysfunction. Neurochem. Int. 2010, 56, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-W.; Liu, D.-X. Humanin Decreases Mitochondrial Membrane Permeability by Inhibiting the Membrane Association and Oligomerization of Bax and Bid Proteins. Acta Pharmacol. Sin. 2018, 39, 1012–1021. [Google Scholar] [CrossRef]
- Romeo, M.; Stravalaci, M.; Beeg, M.; Rossi, A.; Fiordaliso, F.; Corbelli, A.; Salmona, M.; Gobbi, M.; Cagnotto, A.; Diomede, L. Humanin Specifically Interacts with Amyloid-β Oligomers and Counteracts Their in Vivo Toxicity. J. Alzheimer’s Dis. 2017, 57, 857–871. [Google Scholar] [CrossRef]
- Han, K.; Jia, N.; Zhong, Y.; Shang, X. S14G-Humanin Alleviates Insulin Resistance and Increases Autophagy in Neurons of APP/PS1 Transgenic Mouse. J. Cell. Biochem. 2018, 119, 3111–3117. [Google Scholar] [CrossRef]
- Qian, K.; Bao, X.; Li, Y.; Wang, P.; Guo, Q.; Yang, P.; Xu, S.; Yu, F.; Meng, R.; Cheng, Y.; et al. Cholinergic Neuron Targeting Nanosystem Delivering Hybrid Peptide for Combinatorial Mitochondrial Therapy in Alzheimer’s Disease. ACS Nano 2022, 16, 11455–11472. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shi, H.; He, Y.; Yuan, L.; Qu, X.; Zhang, J.; Wang, Z.; Cai, H.; Qi, J. Colivelin Ameliorates Impairments in Cognitive Behaviors and Synaptic Plasticity in APP/PS1 Transgenic Mice. J. Alzheimer’s Dis. 2017, 59, 1067–1078. [Google Scholar] [CrossRef]
- Wu, M.-N.; Zhou, L.-W.; Wang, Z.-J.; Han, W.-N.; Zhang, J.; Liu, X.-J.; Tong, J.-Q.; Qi, J.-S. Colivelin Ameliorates Amyloid β Peptide-Induced Impairments in Spatial Memory, Synaptic Plasticity, and Calcium Homeostasis in Rats. Hippocampus 2015, 25, 363–372. [Google Scholar] [CrossRef]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally Occurring Mitochondrial-Derived Peptides Are Age-Dependent Regulators of Apoptosis, Insulin Sensitivity, and Inflammatory Markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef]
- Wan, W.; Zhang, L.; Lin, Y.; Rao, X.; Wang, X.; Hua, F.; Ying, J. Mitochondria-Derived Peptide MOTS-c: Effects and Mechanisms Related to Stress, Metabolism and Aging. J. Transl. Med. 2023, 21, 36. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018, 28, 516–524.e7. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.X.; Finkel, T. Mitochondria as Intracellular Signaling Platforms in Health and Disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef]
- Jiang, J.; Chang, X.; Nie, Y.; Shen, Y.; Liang, X.; Peng, Y.; Chang, M. Peripheral Administration of a Cell-Penetrating MOTS-c Analogue Enhances Memory and Attenuates Aβ1-42- or LPS-Induced Memory Impairment through Inhibiting Neuroinflammation. ACS Chem. Neurosci. 2021, 12, 1506–1518. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective Effects of Mitophagy Enhancers against Amyloid Beta-Induced Mitochondrial and Synaptic Toxicities in Alzheimer Disease. Hum. Mol. Genet. 2022, 31, 423–439. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal Mitochondrial Dynamics in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective Effects of Reduced Dynamin-Related Protein 1 against Amyloid Beta-Induced Mitochondrial Dysfunction and Synaptic Damage in Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Hemachandra Reddy, P. Corrigendum: Mitochondrial Division Inhibitor 1 Reduces Dynamin-Related Protein 1 and Mitochondrial Fission Activity. Hum. Mol. Genet. 2019, 28, 875–876. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β Induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126 Pt 3, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (MTOR)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a025924. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of MTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-Beta Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, H.; Cen, M.; Tao, Y.; Lai, C.; Tang, Z. Rapamycin Ameliorates Cognitive Impairments and Alzheimer’s Disease-Like Pathology with Restoring Mitochondrial Abnormality in the Hippocampus of Streptozotocin-Induced Diabetic Mice. Neurochem. Res. 2021, 46, 265–275. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular Interplay between Mammalian Target of Rapamycin (MTOR), Amyloid-Beta, and Tau: Effects on Cognitive Impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing Autophagy by Rapamycin before, but Not after, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Shi, Q.; Chang, C.; Saliba, A.; Bhat, M.A. Microglial MTOR Activation Upregulates Trem2 and Enhances β-Amyloid Plaque Clearance in the 5XFAD Alzheimer’s Disease Model. J. Neurosci. 2022, 42, 5294–5313. [Google Scholar] [CrossRef]
- Carosi, J.M.; Sargeant, T.J. Rapamycin and Alzheimer Disease: A Double-Edged Sword? Autophagy 2019, 15, 1460–1462. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Hindle, A.; Kshirsagar, S.; Reddy, P.H. Are Mitophagy Enhancers Therapeutic Targets for Alzheimer’s Disease? Biomed. Pharmacother. 2022, 149, 112918. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The Mitophagy Activator Urolithin A Is Safe and Induces a Molecular Signature of Improved Mitochondrial and Cellular Health in Humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; D’Amico, D.; Andreux, P.A.; Fouassier, A.M.; Blanco-Bose, W.; Evans, M.; Aebischer, P.; Auwerx, J.; Rinsch, C. Urolithin A Improves Muscle Strength, Exercise Performance, and Biomarkers of Mitochondrial Health in a Randomized Trial in Middle-Aged Adults. Cell Rep. Med. 2022, 3, 100633. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A Attenuates Memory Impairment and Neuroinflammation in APP/PS1 Mice. J. Neuroinflammation 2019, 16, 62. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Alvir, R.V.; Pradeepkiran, J.A.; Hindle, A.; Vijayan, M.; Ramasubramaniam, B.; Kumar, S.; Reddy, A.P.; Reddy, P.H. A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease. Cells 2022, 11, 2660. [Google Scholar] [CrossRef]
- Hirano, K.; Fujimaki, M.; Sasazawa, Y.; Yamaguchi, A.; Ishikawa, K.-I.; Miyamoto, K.; Souma, S.; Furuya, N.; Imamichi, Y.; Yamada, D.; et al. Neuroprotective Effects of Memantine via Enhancement of Autophagy. Biochem. Biophys. Res. Commun. 2019, 518, 161–170. [Google Scholar] [CrossRef]
- Jadhav, R.; Kulkarni, Y.A. The Combination of Baicalein and Memantine Reduces Oxidative Stress and Protects against β-Amyloid-Induced Alzheimer’s Disease in Rat Model. Antioxidants 2023, 12, 707. [Google Scholar] [CrossRef]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. Neuroprotective and Cognitive Enhancement Potentials of Baicalin: A Review. Brain Sci. 2018, 8, 104. [Google Scholar] [CrossRef]
- Wang, W.-W.; Han, R.; He, H.-J.; Li, J.; Chen, S.-Y.; Gu, Y.; Xie, C. Administration of Quercetin Improves Mitochondria Quality Control and Protects the Neurons in 6-OHDA-Lesioned Parkinson’s Disease Models. Aging 2021, 13, 11738–11751. [Google Scholar] [CrossRef]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxid. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [PubMed]
- Paula, P.-C.; Angelica Maria, S.-G.; Luis, C.-H.; Gloria Patricia, C.-G. Preventive Effect of Quercetin in a Triple Transgenic Alzheimer’s Disease Mice Model. Molecules 2019, 24, 2287. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Ohta, K. Quercetin Regulates the Integrated Stress Response to Improve Memory. Int. J. Mol. Sci. 2019, 20, 2761. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Y.; Mu, X. Effect of Quercetin on PC12 Alzheimer’s Disease Cell Model Induced by Aβ 25-35 and Its Mechanism Based on Sirtuin1/Nrf2/HO-1 Pathway. BioMed Res. Int. 2020, 2020, 8210578. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Zhuang, X.-X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s Disease Pathology by Mitophagy Inducers Identified via Machine Learning and a Cross-Species Workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef]
- Ni, Y.-Q.; Liu, Y.-S. New Insights into the Roles and Mechanisms of Spermidine in Aging and Age-Related Diseases. Aging Dis. 2021, 12, 1948–1963. [Google Scholar] [CrossRef]
- Wirth, M.; Benson, G.; Schwarz, C.; Köbe, T.; Grittner, U.; Schmitz, D.; Sigrist, S.J.; Bohlken, J.; Stekovic, S.; Madeo, F.; et al. The Effect of Spermidine on Memory Performance in Older Adults at Risk for Dementia: A Randomized Controlled Trial. Cortex J. Devoted Study Nerv. Syst. Behav. 2018, 109, 181–188. [Google Scholar] [CrossRef]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Müller, M.; et al. Dietary Spermidine Improves Cognitive Function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef]
- Jing, Y.-H.; Yan, J.-L.; Wang, Q.-J.; Chen, H.-C.; Ma, X.-Z.; Yin, J.; Gao, L.-P. Spermidine Ameliorates the Neuronal Aging by Improving the Mitochondrial Function in Vitro. Exp. Gerontol. 2018, 108, 77–86. [Google Scholar] [CrossRef]
- Fairley, L.H.; Lejri, I.; Grimm, A.; Eckert, A. Spermidine Rescues Bioenergetic and Mitophagy Deficits Induced by Disease-Associated Tau Protein. Int. J. Mol. Sci. 2023, 24, 5297. [Google Scholar] [CrossRef]
- Makarov, M.; Korkotian, E. Differential Role of Active Compounds in Mitophagy and Related Neurodegenerative Diseases. Toxins 2023, 15, 202. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM Mediates Spermidine-Induced Mitophagy via PINK1 and Parkin Regulation in Human Fibroblasts. Sci. Rep. 2016, 6, 24700. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ Supplementation Normalizes Key Alzheimer’s Features and DNA Damage Responses in a New AD Mouse Model with Introduced DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via CGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.-F.; Lu, Y.-B.; Zhang, W.-P. Nicotinamide Ribose Ameliorates Cognitive Impairment of Aged and Alzheimer’s Disease Model Mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide Riboside Restores Cognition through an Upregulation of Proliferator-Activated Receptor-γ Coactivator 1α Regulated β-Secretase 1 Degradation and Mitochondrial Gene Expression in Alzheimer’s Mouse Models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef]
- Yulug, B.; Altay, O.; Li, X.; Hanoglu, L.; Cankaya, S.; Lam, S.; Velioglu, H.A.; Yang, H.; Coskun, E.; Idil, E.; et al. Combined Metabolic Activators Improve Cognitive Functions in Alzheimer’s Disease Patients: A Randomised, Double-Blinded, Placebo-Controlled Phase-II Trial. Transl. Neurodegener. 2023, 12, 4. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK Inhibitors Upregulate the Neuroprotective Parkin-Mediated Mitophagy Pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef]
- Koch, J.C.; Tönges, L.; Barski, E.; Michel, U.; Bähr, M.; Lingor, P. ROCK2 Is a Major Regulator of Axonal Degeneration, Neuronal Death and Axonal Regeneration in the CNS. Cell Death Dis. 2014, 5, e1225. [Google Scholar] [CrossRef]
- Liu, J.; Gao, H.-Y.; Wang, X.-F. The Role of the Rho/ROCK Signaling Pathway in Inhibiting Axonal Regeneration in the Central Nervous System. Neural Regen. Res. 2015, 10, 1892–1896. [Google Scholar] [CrossRef]
- Saal, K.-A.; Galter, D.; Roeber, S.; Bähr, M.; Tönges, L.; Lingor, P. Altered Expression of Growth Associated Protein-43 and Rho Kinase in Human Patients with Parkinson’s Disease. Brain Pathol. 2017, 27, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tatenhorst, L.; Roser, A.-E.; Saal, K.-A.; Tönges, L.; Lingor, P. ROCK Inhibition in Models of Neurodegeneration and Its Potential for Clinical Translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Stephan, D.A.; Talboom, J.; Corneveaux, J.J.; Reiman, D.M.; Gerber, J.D.; Barnes, C.A.; Alexander, G.E.; Reiman, E.M.; Bimonte-Nelson, H.A. Peripheral Delivery of a ROCK Inhibitor Improves Learning and Working Memory. Behav. Neurosci. 2009, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Shirafuji, N.; Yen, S.-H.; Yoshida, H.; Kanaan, N.M.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Kuriyama, M.; et al. Rho-Kinase ROCK Inhibitors Reduce Oligomeric Tau Protein. Neurobiol. Aging 2020, 89, 41–54. [Google Scholar] [CrossRef]
- Gentry, E.G.; Henderson, B.W.; Arrant, A.E.; Gearing, M.; Feng, Y.; Riddle, N.C.; Herskowitz, J.H. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neurosci. 2016, 36, 1316–1323. [Google Scholar] [CrossRef]
- Hass, D.T.; Barnstable, C.J. Uncoupling Proteins in the Mitochondrial Defense against Oxidative Stress. Prog. Retin. Eye Res. 2021, 83, 100941. [Google Scholar] [CrossRef]
- Kumar, R.; T, A.; Singothu, S.; Singh, S.B.; Bhandari, V. Uncoupling Proteins as a Therapeutic Target for the Development of New Era Drugs against Neurodegenerative Disorder. Biomed. Pharmacother. 2022, 147, 112656. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Cunha, F.M.; Caldeira da Silva, C.C.; Cerqueira, F.M.; Kowaltowski, A.J. Mild Mitochondrial Uncoupling as a Therapeutic Strategy. Curr. Drug Targets 2011, 12, 783–789. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Diano, S.; Horvath, T.L. Mitochondrial Uncoupling Proteins in the CNS: In Support of Function and Survival. Nat. Rev. Neurosci. 2005, 6, 829–840. [Google Scholar] [CrossRef]
- Mehta, S.L.; Li, P.A. Neuroprotective Role of Mitochondrial Uncoupling Protein 2 in Cerebral Stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Ho, P.W.-L.; Ho, J.W.-M.; Liu, H.-F.; So, D.H.-F.; Tse, H.-M.; Chan, K.-H.; Ho, S.-L. Human Neuronal Uncoupling Proteins 4 and 5 (UCP4 and UCP5): Structural Properties, Regulation, and Physiological Role in Protection against Oxidative Stress and Mitochondrial Dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Seiça, R.M.; Moreira, P.I. Uncoupling Protein 2 Inhibition Exacerbates Glucose Fluctuation-Mediated Neuronal Effects. Neurotox. Res. 2018, 33, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Jun, Z.; Ibrahim, M.M.; Dezheng, G.; Bo, Y.; Qiong, W.; Yuan, Z. UCP2 Protects against Amyloid Beta Toxicity and Oxidative Stress in Primary Neuronal Culture. Biomed. Pharmacother. 2015, 74, 211–214. [Google Scholar] [CrossRef]
- Thangavel, R.; Kempuraj, D.; Zaheer, S.; Raikwar, S.; Ahmed, M.E.; Selvakumar, G.P.; Iyer, S.S.; Zaheer, A. Glia Maturation Factor and Mitochondrial Uncoupling Proteins 2 and 4 Expression in the Temporal Cortex of Alzheimer’s Disease Brain. Front. Aging Neurosci. 2017, 9, 150. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; De Nuccio, C.; Minghetti, L.; Visentin, S. The Mitochondrial Uncoupling Protein-2 Is a Master Regulator of Both M1 and M2 Microglial Responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef]
- Rosenberg, N.; Reva, M.; Binda, F.; Restivo, L.; Depierre, P.; Puyal, J.; Briquet, M.; Bernardinelli, Y.; Rocher, A.-B.; Markram, H.; et al. Overexpression of UCP4 in Astrocytic Mitochondria Prevents Multilevel Dysfunctions in a Mouse Model of Alzheimer’s Disease. Glia 2023, 71, 957–973. [Google Scholar] [CrossRef]
- Zorov, D.B.; Andrianova, N.V.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Zorov, S.D.; Zorova, L.D.; Plotnikov, E.Y.; Sukhikh, G.T.; Silachev, D.N. Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sci. 2021, 11, 1050. [Google Scholar] [CrossRef]
- Geisler, J.G.; Marosi, K.; Halpern, J.; Mattson, M.P. DNP, Mitochondrial Uncoupling, and Neuroprotection: A Little Dab’ll Do Ya. Alzheimer’s Dement. 2017, 13, 582–591. [Google Scholar] [CrossRef]
- Aribal, P.; Alver, E.N.; Kaltalioglu, K.; Balabanli, B.; Ebegil, M.; Coskun-Cevher, S. The Relationship between Experimental 2,4-Dinitrophenol Administration and Neurological Oxidative Stress: In Terms of Dose, Time and Gender Differences. Mol. Cell. Biochem. 2023, 478, 1161–1168. [Google Scholar] [CrossRef]
- Geisler, J.G. 2,4 Dinitrophenol as Medicine. Cells 2019, 8, 280. [Google Scholar] [CrossRef]
- De Felice, F.G.; Houzel, J.C.; Garcia-Abreu, J.; Louzada, P.R.; Afonso, R.C.; Meirelles, M.N.; Lent, R.; Neto, V.M.; Ferreira, S.T. Inhibition of Alzheimer’s Disease Beta-Amyloid Aggregation, Neurotoxicity, and in Vivo Deposition by Nitrophenols: Implications for Alzheimer’s Therapy. FASEB J. 2001, 15, 1297–1299. [Google Scholar] [CrossRef]
- Wasilewska-Sampaio, A.P.; Silveira, M.S.; Holub, O.; Goecking, R.; Gomes, F.C.A.; Neto, V.M.; Linden, R.; Ferreira, S.T.; De Felice, F.G. Neuritogenesis and Neuronal Differentiation Promoted by 2,4-Dinitrophenol, a Novel Anti-Amyloidogenic Compound. FASEB J. 2005, 19, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Paula Lima, A.C.; Arriagada, C.; Toro, R.; Cárdenas, A.M.; Caviedes, R.; Ferreira, S.T.; Caviedes, P. Small-Molecule Aggregation Inhibitors Reduce Excess Amyloid in a Trisomy 16 Mouse Cortical Cell Line. Biol. Res. 2008, 41, 129–136. [Google Scholar] [PubMed]
- Liu, D.; Zhang, Y.; Gharavi, R.; Park, H.R.; Lee, J.; Siddiqui, S.; Telljohann, R.; Nassar, M.R.; Cutler, R.G.; Becker, K.G.; et al. The Mitochondrial Uncoupler DNP Triggers Brain Cell MTOR Signaling Network Reprogramming and CREB Pathway Up-Regulation. J. Neurochem. 2015, 134, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, S.; Ali Pour, P.; Kheradvar, A. Prospects of Mitochondrial Transplantation in Clinical Medicine: Aspirations and Challenges. Mitochondrion 2022, 65, 33–44. [Google Scholar] [CrossRef]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal Transfer of Mitochondria between Mammalian Cells: Beyond Co-Culture Approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial Transfer between Cells Can Rescue Aerobic Respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Endo, Y.; Yin, T.; Choudhary, R.C.; Aoki, T.; Nishikimi, M.; Murao, A.; Nakamura, E.; Shoaib, M.; et al. Exogenous Mitochondrial Transplantation Improves Survival and Neurological Outcomes after Resuscitation from Cardiac Arrest. BMC Med. 2023, 21, 56. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Alexander, J.F.; Seua, A.V.; Arroyo, L.D.; Ray, P.R.; Wangzhou, A.; Heiβ-Lückemann, L.; Schedlowski, M.; Price, T.J.; Kavelaars, A.; Heijnen, C.J. Nasal Administration of Mitochondria Reverses Chemotherapy-Induced Cognitive Deficits. Theranostics 2021, 11, 3109–3130. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D. Promising Treatment for Multiple Sclerosis: Mitochondrial Transplantation. Int. J. Mol. Sci. 2022, 23, 2245. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Liou, C.-W.; Chen, S.-D.; Hsu, T.-Y.; Chuang, J.-H.; Wang, P.-W.; Huang, S.-T.; Tiao, M.-M.; Chen, J.-B.; Lin, T.-K.; et al. Mitochondrial Transfer from Wharton’s Jelly-Derived Mesenchymal Stem Cells to Mitochondria-Defective Cells Recaptures Impaired Mitochondrial Function. Mitochondrion 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Shi, X.; Bai, H.; Zhao, M.; Li, X.; Sun, X.; Jiang, H.; Fu, A. Treatment of Acetaminophen-Induced Liver Injury with Exogenous Mitochondria in Mice. Transl. Res. J. Lab. Clin. Med. 2018, 196, 31–41. [Google Scholar] [CrossRef]
- Su, Y.; Zhu, L.; Yu, X.; Cai, L.; Lu, Y.; Zhang, J.; Li, T.; Li, J.; Xia, J.; Xu, F.; et al. Erratum: Mitochondrial Transplantation Attenuates Airway Hyperresponsiveness by Inhibition of Cholinergic Hyperactivity: Erratum. Theranostics 2019, 9, 1385–1386. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.-F.; Mak, J.C.W.; Lian, Q. Mitochondrial Transfer of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke-Induced Damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Nakamura, Y.; Park, J.-H.; Hayakawa, K. Therapeutic Use of Extracellular Mitochondria in CNS Injury and Disease. Exp. Neurol. 2020, 324, 113114. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial Transfer/Transplantation: An Emerging Therapeutic Approach for Multiple Diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Corrigendum: Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 539, 123. [Google Scholar] [CrossRef]
- English, K.; Shepherd, A.; Uzor, N.-E.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes Rescue Neuronal Health after Cisplatin Treatment through Mitochondrial Transfer. Acta Neuropathol. Commun. 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Sun, G.; Bautista Garrido, J.; Obertas, L.; Mobley, A.S.; Ting, S.-M.; Zhao, X.; Aronowski, J. The Mitochondria-Derived Peptide Humanin Improves Recovery from Intracerebral Hemorrhage: Implication of Mitochondria Transfer and Microglia Phenotype Change. J. Neurosci. 2020, 40, 2154–2165. [Google Scholar] [CrossRef]
- Tashiro, R.; Ozaki, D.; Bautista-Garrido, J.; Sun, G.; Obertas, L.; Mobley, A.S.; Kim, G.S.; Aronowski, J.; Jung, J.E. Young Astrocytic Mitochondria Attenuate the Elevated Level of CCL11 in the Aged Mice, Contributing to Cognitive Function Improvement. Int. J. Mol. Sci. 2023, 24, 5187. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Benson, T.; Albensi, B.C. Mitochondrial Transfusion Improves Mitochondrial Function Through Up-Regulation of Mitochondrial Complex II Protein Subunit SDHB in the Hippocampus of Aged Mice. Mol. Neurobiol. 2022, 59, 6009–6017. [Google Scholar] [CrossRef]
- Jia, X.; Wang, Q.; Ji, J.; Lu, W.; Liu, Z.; Tian, H.; Guo, L.; Wang, Y. Mitochondrial Transplantation Ameliorates Hippocampal Damage Following Status Epilepticus. Anim. Models Exp. Med. 2023, 6, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sheng, H.; Liao, L.; Xu, C.; Zhang, A.; Yang, Y.; Zhao, L.; Duan, L.; Chen, H.; Zhang, B. Mesenchymal Stem Cell-Conditioned Medium Improves Mitochondrial Dysfunction and Suppresses Apoptosis in Okadaic Acid-Treated SH-SY5Y Cells by Extracellular Vesicle Mitochondrial Transfer. J. Alzheimer’s Dis. 2020, 78, 1161–1176. [Google Scholar] [CrossRef]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Bobkova, N.V.; Zhdanova, D.Y.; Belosludtseva, N.V.; Penkov, N.V.; Mironova, G.D. Intranasal Administration of Mitochondria Improves Spatial Memory in Olfactory Bulbectomized Mice. Exp. Biol. Med. 2022, 247, 416–425. [Google Scholar] [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, M.I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data from a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Chiang, M.C.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bemis, M.; Desilets, A.R. Role of Medium Chain Triglycerides (Axona®) in the Treatment of Mild to Moderate Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Demen. 2014, 29, 409–414. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, T.; Moreira, P.I.; Cardoso, S. Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease. Biomedicines 2023, 11, 2331. https://doi.org/10.3390/biomedicines11092331
Sousa T, Moreira PI, Cardoso S. Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease. Biomedicines. 2023; 11(9):2331. https://doi.org/10.3390/biomedicines11092331
Chicago/Turabian StyleSousa, Tiago, Paula I. Moreira, and Susana Cardoso. 2023. "Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease" Biomedicines 11, no. 9: 2331. https://doi.org/10.3390/biomedicines11092331
APA StyleSousa, T., Moreira, P. I., & Cardoso, S. (2023). Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease. Biomedicines, 11(9), 2331. https://doi.org/10.3390/biomedicines11092331