1. Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor of childhood, causing about 15% of all pediatric cancer deaths [
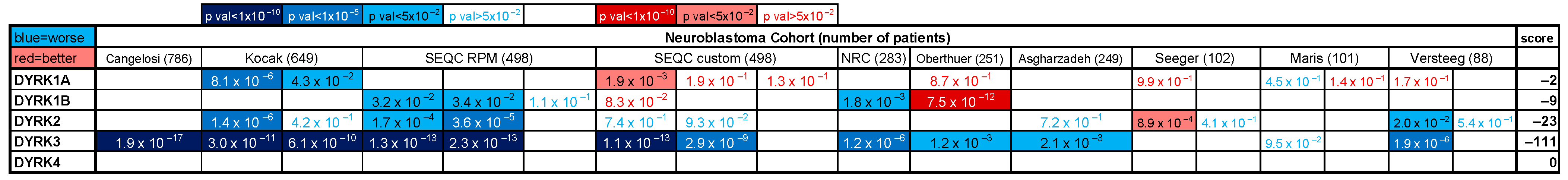
1]. Despite very aggressive multimodal interventions, only ~50% of high-risk NB patients survive, exhibiting serious long-term sequelae from therapy, warranting innovative and improved interventions. In search of potential druggable targets in NB pathogenesis, we performed a systematic analysis of the correlation between expression of the Dual-specificity tYrosine phosphorylation-Regulated Kinase (DYRK) family members and neuroblastoma (NB) patient survival probability. We annotated and ranked the expression of all five
DYRK kinases (
DYRK-1A, 1B, 2, 3, and
4) across 10 publicly available NB patient datasets from the ‘R2: Genomics Analysis and Visualization Platform’ (as previously described [
2]) and observed a significant and robust correlation between
DYRK3 expression and a worse patient survival in 8 of these 10 cohorts, but not for any other family members (
Figure 1). This observation was also true for other neurological tumors (particularly Glioma,
Supplementary Figure S1).
DYRK3 is a largely understudied kinase. Its expression has been associated with a higher aggressiveness of Glioblastoma cells and tumors [
3], although another report suggested it might negatively regulate hepatocellular carcinoma progression [
4]. It is noteworthy that Ivanova et al. [
5] reported a role for DYRK3 as a negative regulator of the hypoxic response and differentiation in NB cells, hence contributing to their aggressive behavior. DYRK3 remains largely understudied, although recent reports from Lucas Pelkmans’ group and others have implicated this kinase as a critical regulator of several biomolecular condensates, such as Stress Granules [
6,
7], centrosomes and the mitotic spindle [
8], endoplasmic reticulum exit sites [
9], or Mediator complex condensates [
10]. Biomolecular condensates—also referred to as membranelles organelles—are sub-cellular compartments that form through liquid–liquid phase separation of RNA and/or proteins that contain intrinsically disordered regions (IDRs) [
11,
12]. Such disordered regions can undergo numerous post-translational modifications, particularly phosphorylation, which regulate their partitioning into these condensates [
13,
14,
15]. We show here that CAMKV is a direct substrate of DYRK3. CAMKV is a protein highly enriched in neuroblastoma cancer cell lines and in healthy neural tissues. Importantly,
CAMKV levels were previously correlated to a worse NB patient survival and its expression is highly associated to that of
MYCN or
MYC in NB cell lines and primary tumor samples [
16].
2. Materials and Methods
2.1. Cell Lines and Culture
The neuroblastoma cell line SJNB-JF-G12 (JF) was originally established in 1979 from a patient with disseminated neuroblastoma and was a kind gift from Dr. Malcom Brenner. The NGP cell line was obtained from DSMZ; SH-SY5Y cells were obtained from ATCC. All cell lines were cultivated at 37 °C with 5% CO2. Cell lines were cultured according to vendors’ recommendations and passaged no more than 8 times. JF cells were grown in RPMI 1640 + 10% h.i. FBS + 4 mM L-Glutamine + pen/strept. NGP cells were grown in DMEM (4.5 g/L glucose) + 10% h.i. FBS + 4 mM L-Glutamine + pen/strept. SH-SY5Y cells were grown in DMEM/F12 (1:1) + 10% h.i. FBS + 4 mM L-Glutamine + penicillin/streptomycin. All cell lines were tested for Mycoplasma every 2 months using a e-MycoTM Mycoplasma PCR Detection Kit (Bulldog Bio #25235), according to the manufacturer’s instructions. All cell lines were authenticated by STR analysis (ATCC).
2.2. Reagents
Harmine and GSK-626616 were obtained from MedChemExpress (Monmouth Junction, NJ, USA) (HY-N0737A and HY-105309, respectively) and resuspended to a 10 mM stock in DMSO for in vitro/cell culture-based assays. DAPI was from Millipore Sigma (Burlington, MA, USA) (#268298). Primary antibodies from Cell Signaling Technology were: mCherry (#43590), β-actin (#4970), vinculin (#13901), Phospho-PRAS40 (Thr246) (#2997), and HA-tag (#3724). Other primary antibodies were: anti-GAPDH from Millipore Sigma (MAB374), anti-DYRK3 from Aviva Systems Biology (San Diego, CA, USA) (ARP30648_P050), anti-CAMKV from Sino Biological (Wayne, PA, USA) (12243-T26). Mouse anti-rabbit (211-035-109) and goat anti-mouse (115-035-146) HRP-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA). Purified human recombinant DYRK3 was obtained from Sino Biological (#10726-H20B). Dynabeads Protein A-magnetic beads for immunoprecipitation were obtained from Invitrogen (Waltham, MA, USA) (#10001D). Lipofectamine 2000 transfection reagent was from Invitrogen (#11668027).
2.3. Western Blot
Western blot analysis was conducted using standard methods as previously described [
17]. Briefly, cells grown to a 60–80% confluency were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Prometheus Protein Biology Products #18-416) supplemented with Protease and Phosphatase Inhibitor Cocktails (Pr/Ph-ICs; Pierce, Thermo Scientific A32955 and A32957). Lysates were sonicated on ice, centrifuged at 15,000×
g at 4 °C for 20 minutes, and the soluble protein fraction was collected. Protein extracts were quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific (Waltham, MA, USA) #23227). A total of 30–50 μg of proteins were separated via SDS-PAGE using Novex™ WedgeWell™ 4–20%, Tris-Glycine Mini Protein Gels (Invitrogen, Thermo Scientific XP04202BOX) and blotted onto a PVDF membrane using an iBlot transfer system and transfer stacks (Invitrogen, Thermo Scientific IB401001). Proteins were detected using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific 34580). A ChemiDoc MP Imaging System (Bio-Rad, Tokyo, Japan) was used for chemiluminescent detection and analysis.
2.4. Immunoprecipitation and In Vitro Kinase Assay
For soluble cell lysates, cells were washed twice in PBS and lysed in IP buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% NP-40, 10% glycerol, Pr/Ph-ICs). Lysates were clarified by centrifugation and protein quantification with the BCA assay kit (Pierce). One mg of total protein/sample was incubated for 6 h at 4 °C with protein A/G-magnetic beads (Dynabeads, Invitrogen, ThermoFisher Scientific (Vilnius, Lithuania)) prebound with 5 μg of anti-mCherry antibody, and then beads were washed with IP buffer. Finally, both lysates (input) and the immuno-precipitates were resuspended in 5× LB and analyzed by Western blotting. For in vitro kinase (IVK) from immunocomplexes, cell lysates were prepared in IP lysis buffer (50 mM HEPES [pH 7.4], 75 mM NaCl, 1 mM EDTA, 1% NP-40, Pr/Ph-ICs). Cell lysates were incubated overnight at 4 °C with anti-mCherry bound to protein A-magnetic beads (Dynabeads, Invitrogen). Immunocomplexes were washed 4 times with IP and twice time with kinase buffer (25 mM Hepes pH 7.4, 5 mM MgCl2, 5 mM MnCl2, 0.5 mM DTT). Anti-mCherry immuno-complexes were split into 3 aliquots: 5% for Western blotting and 2 aliquots of 45% for IVK assay, with and without human recombinant DYRK3. Immunocomplexes were incubated for 20 min at 30 °C in 30 μL of kinase buffer with a final concentration of 50 μM ATP and [γ-32P] ATP (1 × 10−2 μCi/pmol, PerkinElmer (Shelton, CT, USA)). Reactions were stopped by adding 5× LB, and samples were resolved by SDS-PAGE and then stained with Coomassie blue. Incorporation of 32P was detected by autoradiography of dried gels.
2.5. Orthotopic Xenograft Renal Capsule Injection
One million Tet-shDYRK3 JF cells suspended in 0.1 mL of PBS were surgically implanted in the left renal capsule of NSG immunodeficient mice (The Jackson Laboratory (Bar Harbor, ME, USA) #005557), as previously described [
18]. After 7 days, mice were randomly divided into ‘control’ (untreated) group vs. ‘doxy’ group, receiving 1 mg/mL doxycycline in the drinking water for 3 additional weeks. The body weight of mice was monitored weekly. At the end of the treatment, all mice were euthanized. Tumors and the right kidneys (control) were dissected and weighed. All procedures were approved by the Institutional Animal Care and Utilization Committee (IACUC) at UMass Chan Medical School and according to our IACUC approved protocol (A3306-01).
2.6. RT-qPCR
Total RNA was extracted from cells using the Quick-RNA MiniPrep Kit (Zymo Research (Irvine, CA, USA) #11-327) following the manufacturer’s protocol. cDNA was synthesized from 0.5 ug/sample of total RNA with the ABScript II RT Mix (ABclonal (Woburn, MA, USA) RM21452) according to the manufacturer’s instruction. The cDNA was amplified in 96-well reaction plates with a Universal SYBR Green Fast qPCR Mix (ABclonal RM21203) on a QuantStudio 3 real-time PCR system (Applied Biosystems (Waltham, MA, USA), Thermo Fisher Scientific). The sequences of forward and reverse primers are available in the ‘
Supplementary Material’ section. The relative level of target transcripts was calculated from triplicate samples after normalization against human TBP and/or GAPDH transcripts. Dissociation curve analysis was performed after PCR amplification to confirm primers’ specificity. Relative mRNA expression was calculated using the ΔΔCT method.
2.7. Cell Proliferation Analysis
Cell proliferation of indicated conditions (untreated vs. shRNA-expressing cells) was measured as the relative whole-well confluency of 96-well culture plates using a Celigo Imaging Cytometer (Nexcelom Bioscience LLC, Lawrence, MA, USA). Briefly, ~1000 cells/well were plated on day-1 and incubated for 24 h to allow the cells to attach and recover. The following day each well was imaged and analyzed for relative confluence (day 0). After imaging, cells were treated with vehicle (DMSO, ‘untreated control’) or the indicated final concentrations of Harmine or Doxycycline for the indicated times. Relative confluence was subsequently analyzed every 24/48 h until untreated control wells reached 70–90% confluency. Each condition was performed in 3–6 replicates per experiment. Medium +/− treatment was changed every 48–72 h. Cell proliferation was plotted as the time-dependent change of the average relative confluency for each condition using GraphPad Prism 8 software.
2.8. Immunofluorescence Analysis
Standard immunofluorescence techniques were used as recommended in the Cell Signaling Technology protocols webpage. Briefly, cells were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature or in 100% ice-cold methanol for 15 min at 4 °C (for anti-CAMKV), washed 2× in PBS (5 min/wash), and 2× with 0.2% Triton X-100 in PBS (‘PBS-T’). Cells were blocked in 2% BSA-PBS-T (blocking buffer) for 60 min at 4 °C and incubated with primary antibodies diluted in blocking buffer overnight at 4 °C. Then, 4× washes in PBS and 1 h incubation with secondary antibody Alexa 488 goat anti-rabbit (Invitrogen) followed by 4× additional washes in PBS. Images were obtained in an Echo Revolve fluorescence microscope (BICO (San Diego, CA)) or in a Zeiss LSM700 (Peabody, MA) confocal microscope (UMass Chan Medical School). Images of mCherry-expressing constructs were obtained from in vivo microscopy with the Echo Revolve fluorescence microscope.
2.9. Lentivirus Preparation and Infection
HEK-293T cells were maintained at 37 °C in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% FCS and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin). Cells were transfected with pVSV-G [
19] and pCMV∆R8.91 [
20], together with the pLKO.1-puro non-targeting vector (Sigma Mission clone SHC016; ‘shControl’) or the pLKO.1-puro-shRNA vectors to target DYRK3 or CAMKV (Sigma Mission clone numbers available in the ‘
Supplementary Material’ section, obtained from the UMass Chan Medical School RNAi core facility) using Lipofectamine
TM 2000 reagent (Invitrogen) as recommended by the fabricant, and following the recommendations of The RNAi Consortium (TRC) laboratory protocols with slight modifications. Twelve hours after transfection the medium was replaced by DMEM, supplemented with 30% FCS and antibiotics. Cell supernatants were harvested every 24 h, replaced with fresh medium, and stored at 4 °C until collection of the last harvest (at 72 h). At this point, the consecutive harvests were pooled, filtered through 0.45 μm filters, and split in 3–5 mL aliquots, which were stored at −80 °C. NB cells were infected with shControl or shRNA lentiviral particles by adding a 1:1 mix of medium:viral supernatant for 24–48 h. Puromycin selection (2 μg/mL) was applied for 2–3 days and always compared to non-transduced control cells, which generally died within the first 24 h. Target downregulation was confirmed by Western blot and/or RT-qPCR. For mCherry overexpressing constructs the same lentiviral production strategy was followed, using a lentiviral expression construct instead of the pLKO.1-puro shRNA vectors. pLV-mCherry-CAMKV (WT) under the medium-strength promoter EFS (Human eukaryotic translation elongation factor 1 α1 short form) was custom designed and ordered from VectorBuilder. ‘mCherry-only’ and ‘mCherry-CAMKV-∆IDR’ variants were cloned from the original pLV-mCherry-CAMKV construct by standard molecular biology techniques. For the tetracycline-inducible shDYRK3 system, the Tet-pLKO-puro vector was obtained from Addgene (#21915) and cloning of the shDYRK3-2 hairpin (TRCN0000000647) was performed as recommended in the Tet-pLKO manual (available at
www.addgene.org). Cells expressing mCherry were purified by FACS at the UMass Chan FACS core facility. All constructs were confirmed by Sanger sequencing.
2.10. Statistical Analyses
All quantitative data points represent the mean of three independent experiments performed in 3 or more replicates with standard deviation (S.D). Statistical analysis was performed using a t-test or two-way ANOVA (GraphPad Software, Inc., La Jolla, CA, USA).
3. Results
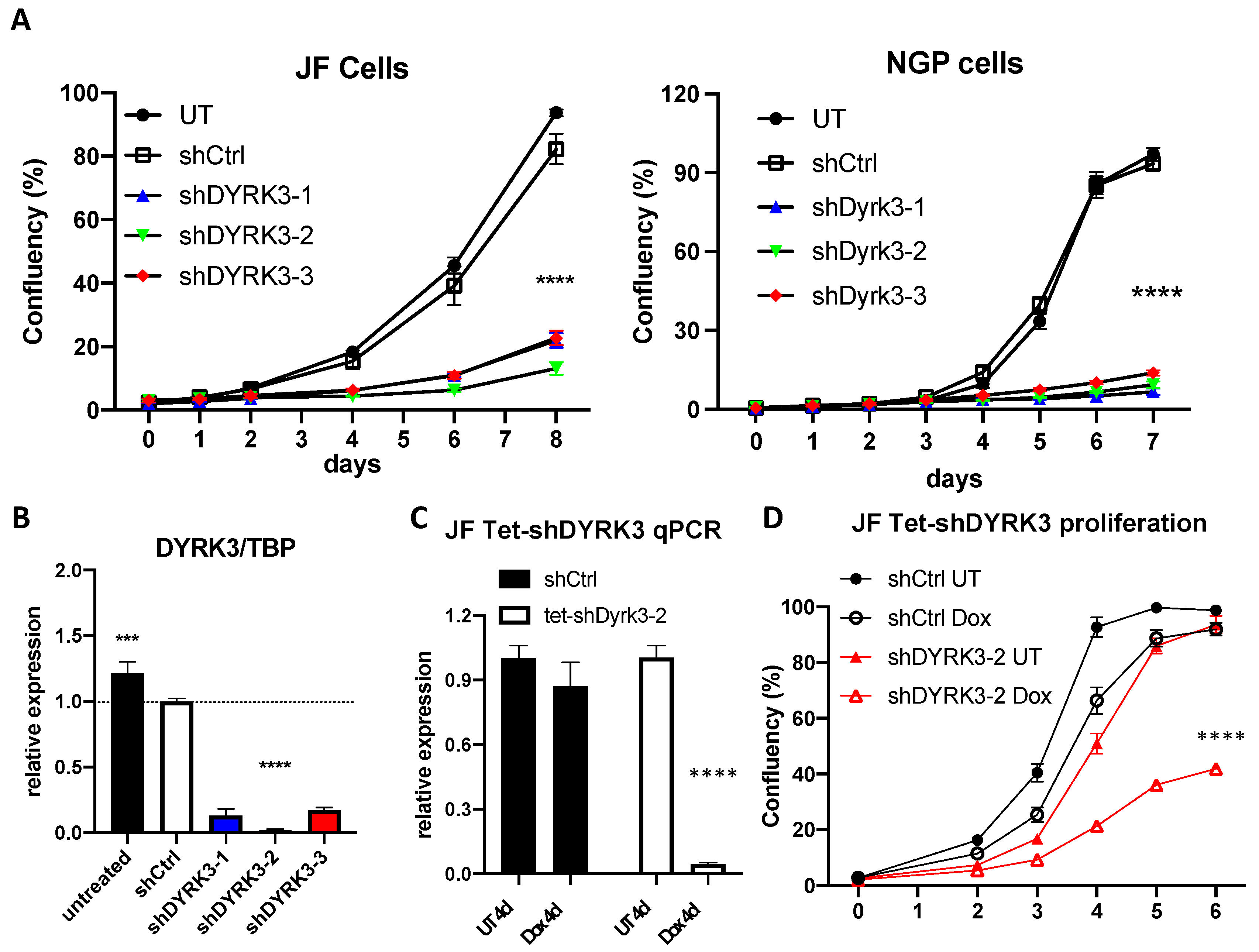
To investigate in more detail the relation between DYRK3 levels and neuroblastoma pathophysiology, we knocked down its expression by means of three specific shRNA-expressing lentiviral clones (shDYRK3-1/2/3) vs. a non-targeting control (shCtrl) in two different NB cell lines. In this way, we observed a very striking impairment in NB cell proliferation (
Figure 2A). Efficient and specific downregulation of
DYRK3 was confirmed by quantitative Real-Time PCR (RT-qPCR;
Figure 2B and
Figure S2), demonstrating a critical role for DYRK3 in neuroblastoma cell homeostasis. To further examine DYRK3 implications in NB tumorigenesis, we established a tetracycline-inducible shDYRK3 expression system (Tet-shDYRK3 JF cells). Validation of this paradigm by RT-qPCR and Western blot (WB) confirmed efficient downregulation of DYRK3 upon doxycycline treatment (
Figure 2C and
Figure S3), again resulting in significantly impaired cell proliferation (
Figure 2D). Treatment of JF NB cells with low doses (1 μM) of Harmine, a pan-DYRK inhibitor known to block DYRK3, resulted in a moderate but significant inhibition of cell proliferation (
Supplementary Figure S4). At higher doses (10 μM) all cells died within 48 h, suggesting a non-specific cytotoxic effect.
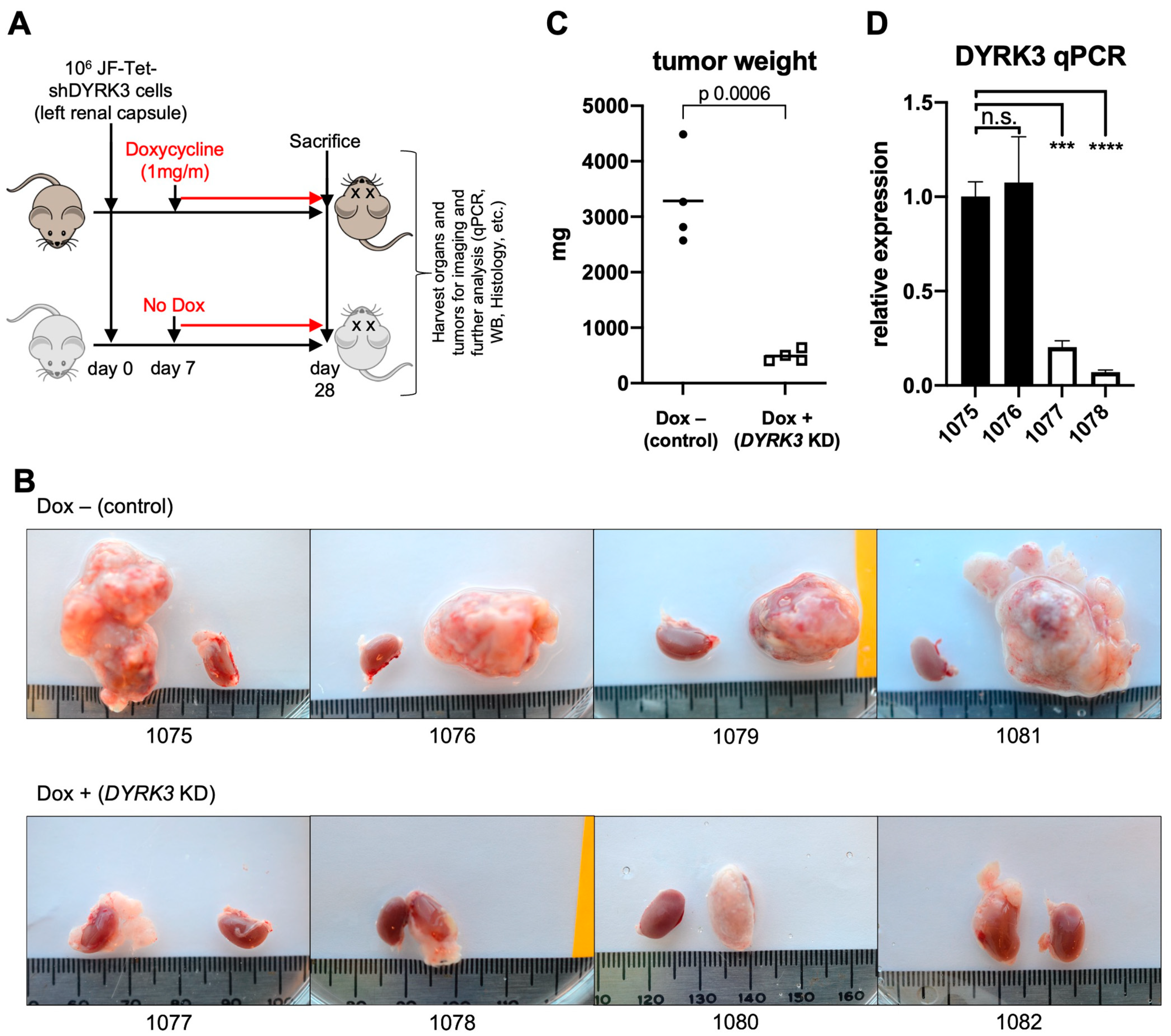
To assess the direct role of DYRK3 in NB tumorigenesis in vivo, Tet-shDYRK3 JF cells were orthotopically injected into the renal capsule of immuno-compromised NSG
TM recipient mice. Seven days post-surgery (7 dps), mice were randomly divided into a ‘control’ (untreated) group vs. ‘doxy’ group, receiving 1 mg/mL doxycycline in the drinking water for three additional weeks (
Figure 3A). Importantly, all mice in the control group developed large tumors, while the doxy group tumors were dramatically smaller (
Figure 3B,C). As a control, the contralateral healthy kidneys showed no significant weight differences (
Supplementary Figure S5). RT-qPCR analysis from tumor RNA samples reflected the efficient downregulation of
DYRK3 by doxycycline treatment (
Figure 3D), thus confirming a critical and previously unrecognized role for this kinase in NB cell proliferation and in vivo tumor growth. As mentioned above, Ivanova et al. [
5] reported a role for DYRK3 as a negative regulator of the hypoxic response and differentiation in NB cells, hence contributing to their aggressive behavior. Surprisingly, this work was entirely based on DYRK3 endogenous or ectopic expression, but no pharmacological inhibition or downregulation was performed, and it did not examine potential effects on NB cell proliferation. Hence, our robust preliminary results are not in disagreement with such a function in a hypoxic setting.
In search of a mechanistic target, we explored the literature for known DYRK3 substrates that could have a specific role in NB tumorigenesis. Wippich et al. [
6] performed an in vitro kinase substrate identification screen using protein microarrays in the presence of wild-type (WT) recombinant DYRK3 vs. a kinase dead mutant (K128M) as a negative control. Among the 26 protein target hits phosphorylated only by WT DYRK3, they found FIP1L1, AKT1S1, and CAMKV as the top three candidates by average Z-score. The authors went on to characterize AKT1S1 (PRAS40) as a novel phosphorylation substrate of DYRK3 in stress granule biology.
We thus became interested in CAMKV, which was previously correlated to a worse NB patient survival and whose mRNA expression is highly associated to that of
MYCN or
MYC in NB cell lines and primary tumor samples [
16]. The Calmodulin Kinase-like Vesicle-associated (CAMKV) protein is a member of the Ca
2+/calmodulin-dependent protein kinase family, highly enriched in brain and endocrine tissues [
21]. Due to a lack of structural conservation in key residues, CAMKV is predicted to have impaired kinase activity, although experimental validation of this idea awaits full validation. In agreement with a previous report [
16], we observed that
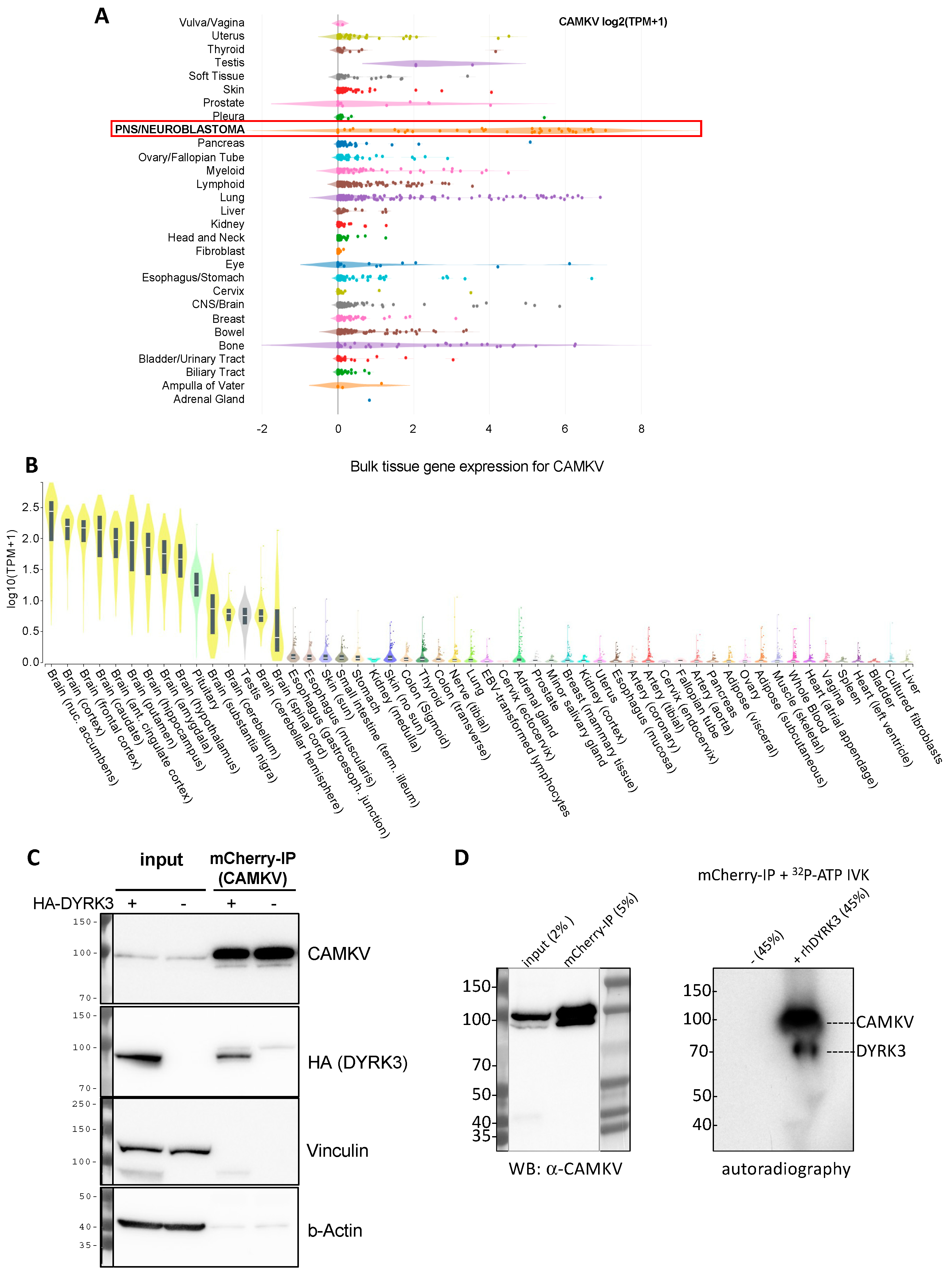
CAMKV is highly enriched in neuroblastoma cancer cell lines (
Figure 4A) and in healthy neural tissues (
Figure 4B).
We thus decided to explore in more detail the potential relation between DYRK3 and CAMKV in NB cell homeostasis. To corroborate that CAMKV indeed can interact with DYRK3, we used the NGP NB cell line transduced with a lentiviral vector to stably over-express an mCherry-CAMKV fusion protein. These cells were transiently transfected with an HA-tagged DYRK3 WT (HA-DYRK3) construct or an empty vector as control, and the corresponding cell lysates were subjected to immuno-precipitation (IP) with an mCherry-specific antibody. Western blot analysis of these immuno-precipitates revealed a clear anti-HA signal in the HA-DYRK3-tranfected sample, but not in the control, demonstrating DYRK3 coprecipitation with CAMKV (
Figure 4C). Finally, mCherry immuno-precipitates from NGP mCherry-CAMKV cells were subjected to in vitro kinase assays (IVKs) with radioactive [γ-
32P]-ATP, in the absence or presence of recombinant human DYRK3. This approach confirmed the direct in vitro phosphorylation of CAMKV by DYRK3 (
Figure 4D).
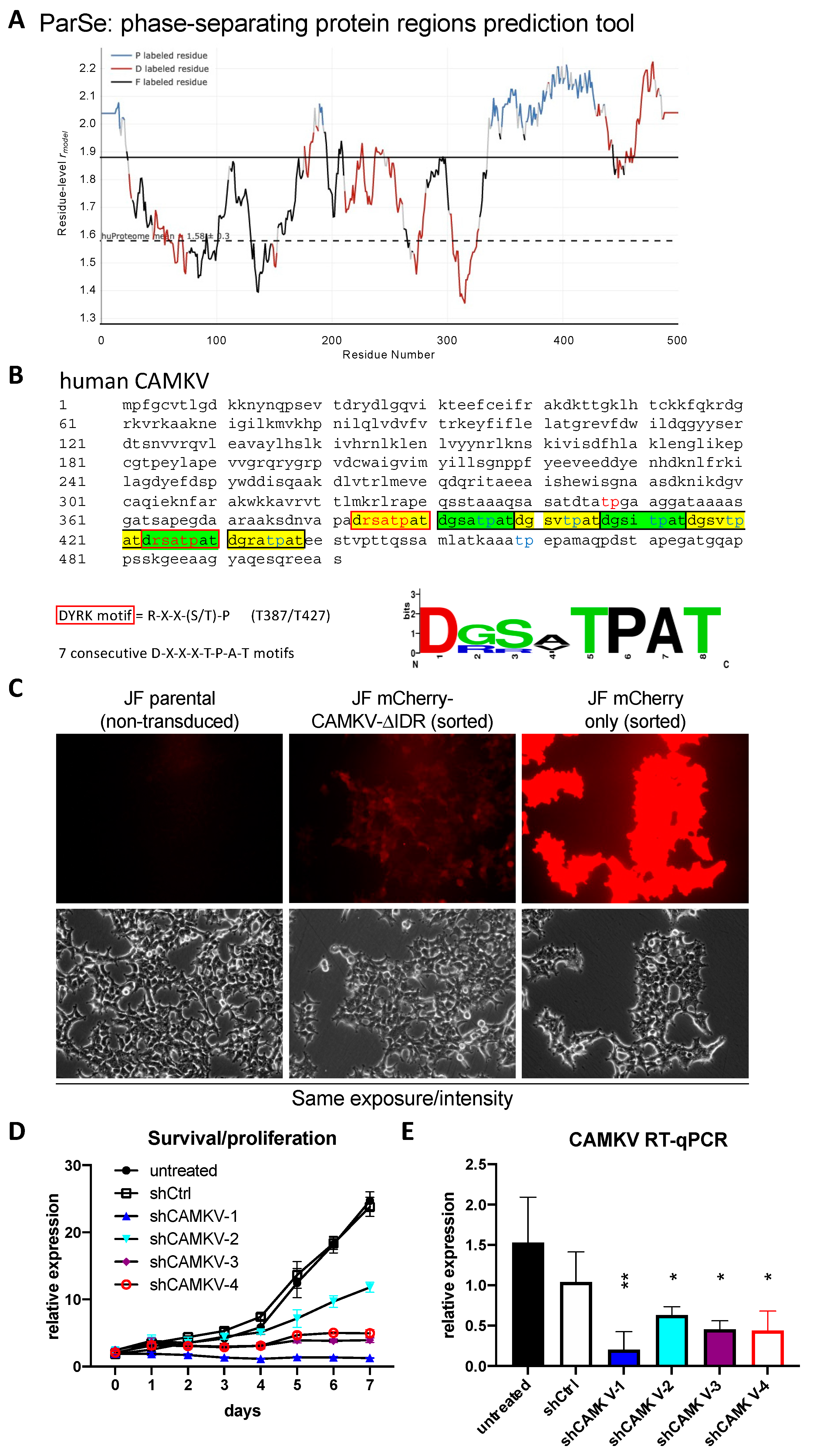
Given the known roles for DYRK3 as a critical regulator of the liquid–liquid phase separation (LLPS) behavior of its substrates, we then focused on CAMKV’s protein sequence. Barylko et al. [
21] recently reported a predicted intrinsically disordered region of ~200 amino acids in CAMKV’s C-terminal half. We employed bioinformatic tools for the prediction of LLPS formation —ParSe (phase-separating protein regions prediction tool;
Figure 5A)— or for prediction of disordered regions —PONDR
® (Predictor of Natural Disordered Regions) and PrDOS (Protein DisOrder prediction System;
Supplementary Figure S6)—, which further confirmed the presence of a highly disordered C-terminal region likely capable of undergoing phase separation in a phosphorylation-regulated manner. Upon a deeper look, we identified seven tandem repeats of an octapeptide motif (D-X-X-X-T-P-A-T), including two canonical and five highly related DYRK phosphorylation motifs (
Figure 5B). In fact, in the original characterization of rat Camkv, Godbout et al. [
22] briefly described such region as a potential ‘PEST sequence’, a motif known to act as a signal for degradation in proteins with a short half-life [
23,
24]. In this context, our overexpression experiments with the lentivirally encoded mCherry-CAMKV WT construct suggested CAMKV as a very stable protein (see below), as demonstrated by the high and constant expression of mCherry-CAMKV across many passages with no loss (but rather increase) of the signal with time. Furthermore, in an attempt to characterize the role of these unique tandem motifs, we generated an overexpression mutant version, ‘mCherry-CAMKV-∆IDR’, lacking the 56 amino acids corresponding to the seven tandem octapeptide repeats. Surprisingly, the mutant variant exhibited a very low to null expression, suggesting that the resulting protein was very unstable (
Figure 5C), and confirming that the seven tandem octapeptide repeats are needed for expression or stabilization—and likely function—of CAMKV, and not acting as a canonical PEST sequence. Interestingly, shRNA-mediated downregulation of
CAMKV by shRNA lentiviral vectors also resulted in a striking impairment of NB cell proliferation (
Figure 5D). Of note, the level of
CAMKV downregulation by different shRNA efficiencies (
Figure 5E) was nicely correlated to the degree of proliferation potential of these cells (
Figure 4D), suggesting that CAMKV is required for NB cell proliferation or survival in an expression level-dependent fashion.
To examine whether CAMKV might form or localize to membraneless organelles, we analyzed our mCherry-CAMKV overexpressing cells by fluorescence microscopy. CAMKV was originally predicted to lack a kinase activity and associated with neuronal vesicles of the rat cortex [
22]. More recent work in mouse models implies a role in activity-dependent bulk endocytosis during the recycling of synaptic vesicles [
25], while Liang et al. [
26] suggested the co-localization of Camkv with postsynaptic scaffold protein PSD-95 puncta, where it may be required for the activity-dependent maintenance of dendritic spines. Nevertheless, in those immunofluorescent staining images, Camkv was not specifically localized to the dendritic spines, but rather homogeneously distributed all along the neuron. Additionally, Barylko et al. [
21] found that murine Camkv can undergo palmitoylation on its N-terminal end, and that this modification was necessary for plasma membrane localization of a Camkv-EGFP (C-terminal) fusion construct. Interestingly, the authors noted that an N-terminal fusion construct (EGFP-Camkv) did not localize to the membrane, but had a homogeneous cytosolic distribution. As for human CAMKV, Sussman et al. [
16] also suggested a membrane localization in NB cell lines, although their data were largely inconclusive.
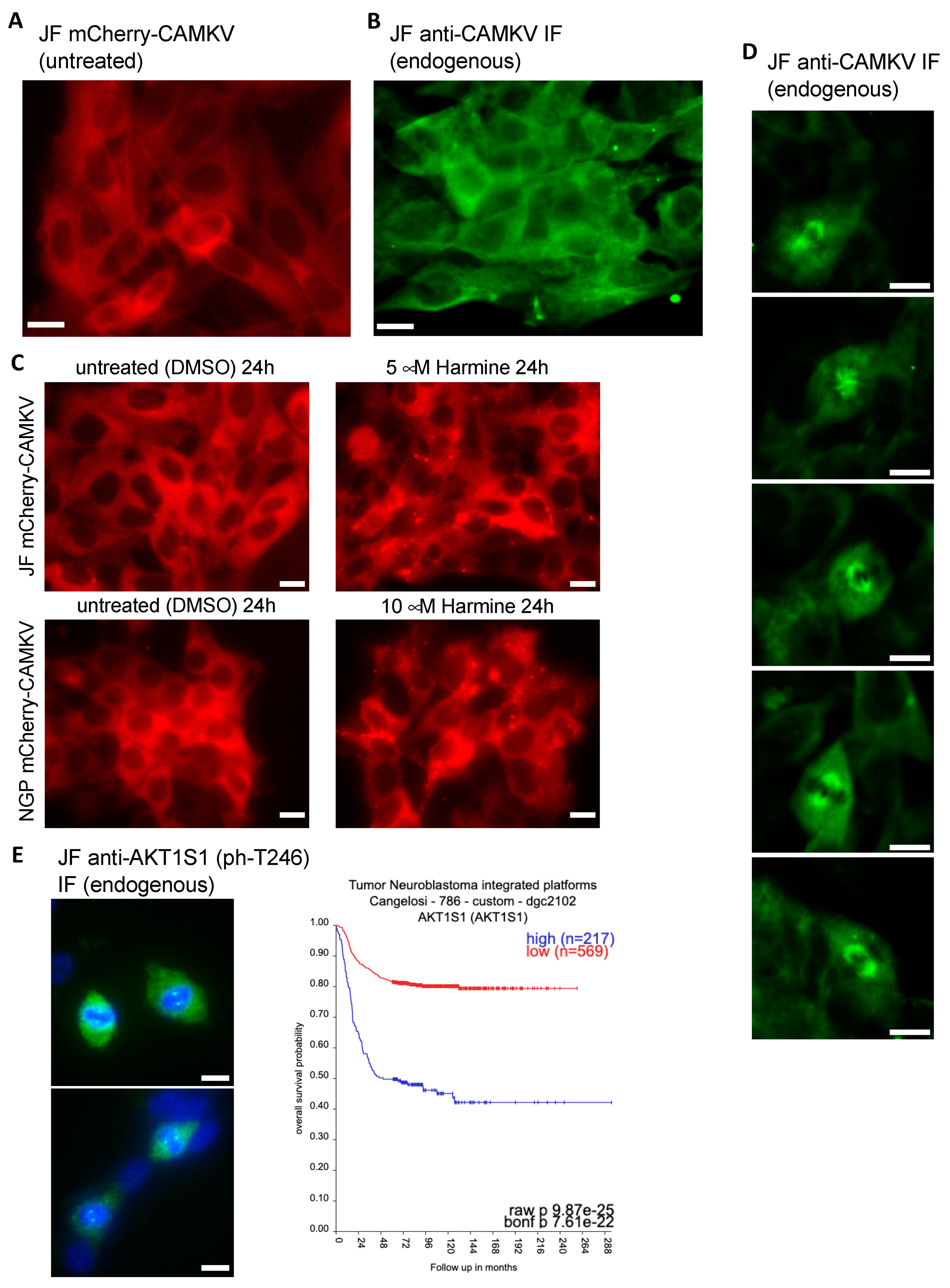
In our hands, ectopic expression of mCherry-CAMKV in NB cell lines showed a clear homogeneous cytosolic pattern (
Figure 6A), consistent with that reported by Barylko et al. [
21] for EGFP-Camkv. More importantly, immuno-fluorescent staining of endogenous CAMKV confirmed a similar cytoplasmic distribution in interphasic NB cells (
Figure 6B). Since inhibition of DYRK3 has been shown to affect the organization of several biomolecular condensates [
6,
7,
8,
9,
10], we treated mCherry-CAMKV JF and NGP cells with Harmine, a pan-DYRK inhibitor known to block DYRK3. Interestingly, this treatment resulted in the relocalization of mCherry-CAMKV into numerous aggregates (
Figure 6C and
Figure S7) with a very dynamic behavior (
Supplementary videos S1 and S2). Treatment of these cells with an unrelated DYRK3 inhibitor, GSK-626616, showed similar effects (
Supplementary Figure S8). These results are consistent with previous observations in other biomolecular condensates upon DYRK3 inhibition [
6,
8,
9], and suggest that CAMKV might indeed undergo liquid–liquid phase separation in a DYRK3-regulated fashion.
It is noteworthy that we failed to observe aggregation of endogenous CAMKV upon Harmine treatment. This might suggest that the mCherry-CAMKV aggregates are a result, at least in part, from its non-physiological over-expression, or that endogenous CAMKV aggregates are sensitive to the harsh methanol fixation used for this staining. Importantly, in our immuno-fluorescence staining analyses of endogenous CAMKV we noticed that virtually every NB cell undergoing cell division displayed a considerably higher anti-CAMKV signal as compared to neighboring interphasic cells. Surprisingly, this signal corresponded to a very clear staining of the mitotic spindle (
Figure 6D and
Supplementary Figure S9), a transitory structure fundamental for the progression of the cell cycle, and whose organization is governed by liquid–liquid phase separation [
27,
28,
29]. As mentioned above, Rai et al. [
8] reported DYRK3 colocalization with Pericentrin in the mitotic spindle poles, further supporting a potential direct role in the regulation of CAMKV function during cell division. Interestingly, we also observed a clear localization in the mitotic spindle poles for endogenous phospho-Thr246 AKT1S1 in dividing JF cells (
Figure 6E left panel). As also mentioned above, Wippich et al. [
6] characterized the direct phosphorylation of Thr246-AKT1S1 by DYRK3, although a function for this signaling module in the regulation of the mitotic spindle has never been reported. Importantly, we further noticed that
AKT1S1 expression is also highly correlated to a worse NB patient outcome (
Figure 6E right panel), as has been suggested for several other cancer types [
30]. This observation supports the idea that DYRK3 might act as a central orchestrator of the mitotic spindle organization by recruiting and/or phosphorylating CAMKV and AKT1S1. Therefore, we propose that downregulation or pharmacologic inhibition of DYRK3, CAMKV, and AKT1S1 in NB cells results in an impairment of the mitotic spindle organization and subsequent exit from the cell cycle, a function that will be the focus of future work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





