
Impact of Obesity and Lysosomal Dysfunction on Chemoresistance in Ovarian Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
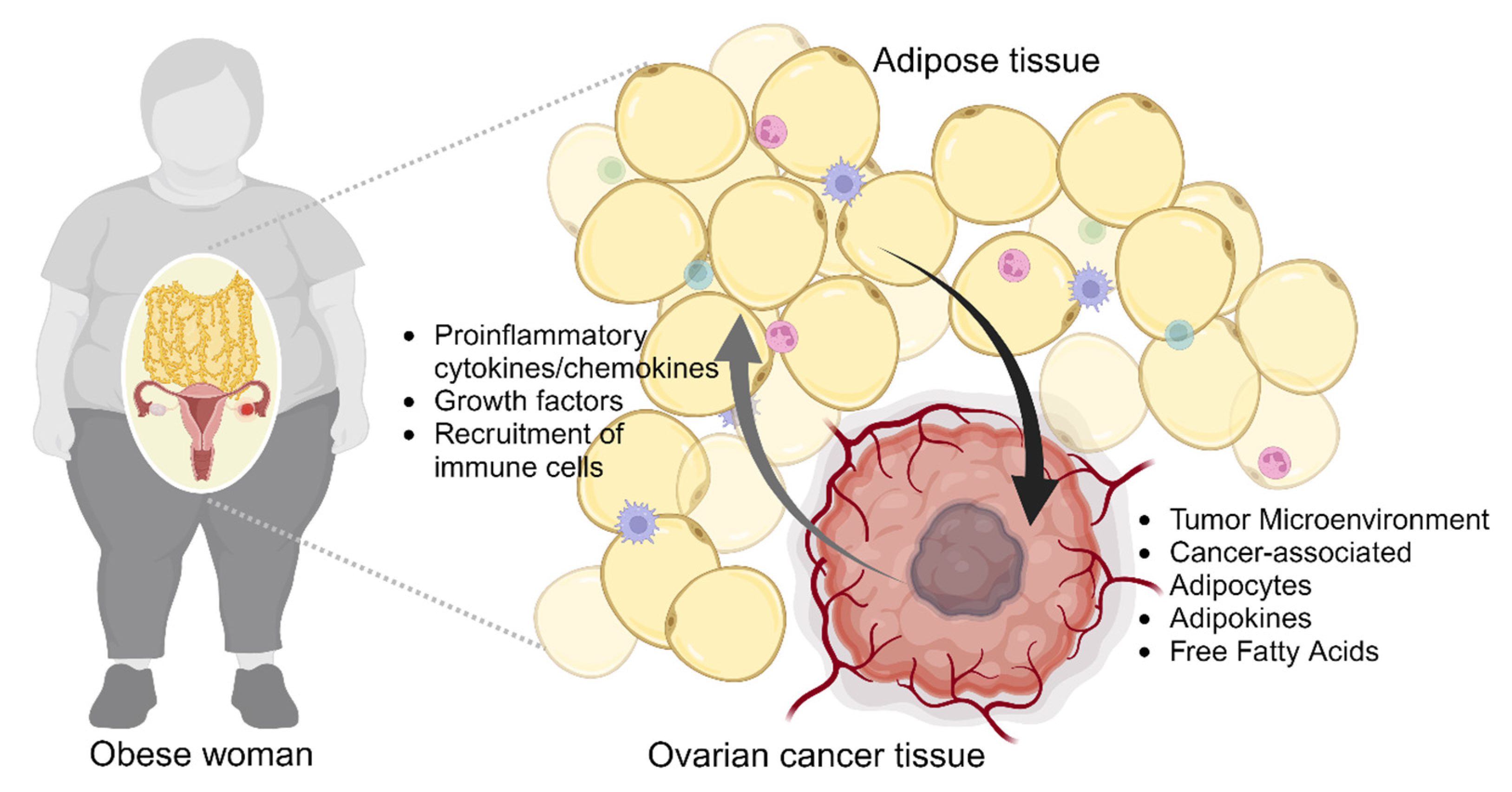
2. Obesity and Ovarian Cancer
2.1. Adiposity and Ovarian Cancer Cells
2.2. Obesity-Induced Remodeling of Tumor Microenvironment
2.3. Lipid Metabolism in Ovarian Cancer
3. Lysosomal Dysfunction in Obesity and Cancer
3.1. Lysosomal Dysfunction in Diseases
3.2. Lysosomal Dysfunction in Diseases
3.2.1. Obesity
3.2.2. Cancer
3.3. Lysosomal Calcium Regulation in Adipocytes and Ovarian Cancer Cells
3.4. Drug Sequestration by Dysfunctional Lysosomes in Cancer
3.4.1. Sequestration of Anticancer Agents by Lysosomal Weak Bases
3.4.2. Anticancer Drug Sequestration via ATP-Binding Cassette Transporters
4. Conclusions
Funding
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef]
- Christie, E.L.; Bowtell, D.D.L. Acquired chemotherapy resistance in ovarian cancer. Ann. Oncol. 2017, 28, viii13–viii15. [Google Scholar] [CrossRef]
- Cooke, S.L.; Brenton, J.D. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011, 12, 1169–1174. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Horowitz, M.; Esakov, E.; Rose, P.; Reizes, O. Signaling within the epithelial ovarian cancer tumor microenvironment: The challenge of tumor heterogeneity. Ann. Transl. Med. 2020, 8, 905. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, T.T.; Zhao, J.J.; Qi, S.F.; Du, P.; Liu, D.W.; Tian, Q.B. The association between overweight, obesity and ovarian cancer: A meta-analysis. Jpn. J. Clin. Oncol. 2015, 45, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, D.L.; Chen, Z.Z.; Gou, B.F. Associations of body mass index with cancer incidence among populations, genders, and menopausal status: A systematic review and meta-analysis. Cancer Epidemiol. 2016, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leitzmann, M.F.; Koebnick, C.; Danforth, K.N.; Brinton, L.A.; Moore, S.C.; Hollenbeck, A.R.; Schatzkin, A.; Lacey, J.V., Jr. Body mass index and risk of ovarian cancer. Cancer 2009, 115, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Poorolajal, J.; Jenabi, E.; Masoumi, S.Z. Body mass index effects on risk of ovarian cancer: A meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 7665–7671. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Calle, E.E.; Fakhrabadi-Shokoohi, D.; Jacobs, E.J.; Thun, M.J. Body mass index, height, and the risk of ovarian cancer mortality in a prospective cohort of postmenopausal women. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 822–828. [Google Scholar]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Chiang, C.Y.; Daifotis, H.A.; Nieman, K.M.; Fahrmann, J.F.; Lastra, R.R.; Romero, I.L.; Fiehn, O.; Lengyel, E. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res. 2020, 80, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zaman, M.M.; Vlasakov, I.; Roy, R.; Huang, L.; Martin, C.R.; Freedman, S.D.; Serhan, C.N.; Moses, M.A. Adipocytes promote ovarian cancer chemoresistance. Sci. Rep. 2019, 9, 13316. [Google Scholar] [CrossRef] [PubMed]
- Chehade, H.; Tedja, R.; Ramos, H.; Bawa, T.S.; Adzibolosu, N.; Gogoi, R.; Mor, G.; Alvero, A.B. Regulatory Role of the Adipose Microenvironment on Ovarian Cancer Progression. Cancers 2022, 14, 2267. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, Y.; Yang, P.; Chen, Y.; Wei, L.; Yu, T.; Xia, J.; Ruan, X.Z.; Zhao, L.; Chen, Y. Obesity induces preadipocyte CD36 expression promoting inflammation via the disruption of lysosomal calcium homeostasis and lysosome function. eBioMedicine 2020, 56, 102797. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W., Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, G.; Kim, H.; Song, Y.S.; Jung, J. Modulation of Cisplatin Sensitivity through TRPML1-Mediated Lysosomal Exocytosis in Ovarian Cancer Cells: A Comprehensive Metabolomic Approach. Cells 2024, 13, 115. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Huang, T. Obesity and Ovarian Cancer. Recent Results Cancer Res. 2016, 208, 155–176. [Google Scholar] [CrossRef]
- Foong, K.W.; Bolton, H. Obesity and ovarian cancer risk: A systematic review. Post Reprod. Health 2017, 23, 183–198. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Mentoor, I.; Engelbrecht, A.M.; Nell, T. Fatty acids: Adiposity and breast cancer chemotherapy, a bad synergy? Prostaglandins Leukot. Essent. Fatty Acids 2019, 140, 18–33. [Google Scholar] [CrossRef]
- Iwase, T.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Sakakibara, J.; Hayama, S.; Ishigami, E.; Masuda, T.; Miyazaki, M. Impact of body fat distribution on neoadjuvant chemotherapy outcomes in advanced breast cancer patients. Cancer Med. 2016, 5, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, J.G.; Balevic, S.; Sinha, J.; Perrin, E.M.; Wang, J.; Edginton, A.N.; Gonzalez, D. Characterizing Pharmacokinetics in Children with Obesity-Physiological, Drug, Patient, and Methodological Considerations. Front. Pharmacol. 2022, 13, 818726. [Google Scholar] [CrossRef]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus white adipose tissue: A mini-review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Mora, B.C.; Venkateswaran, V. Diet, obesity, and cancer progression: Are adipocytes the link? Lipid Insights 2013, 6, 37–45. [Google Scholar] [CrossRef]
- Lashinger, L.M.; Rossi, E.L.; Hursting, S.D. Obesity and resistance to cancer chemotherapy: Interacting roles of inflammation and metabolic dysregulation. Clin. Pharmacol. Ther. 2014, 96, 458–463. [Google Scholar] [CrossRef]
- Delort, L.; Kwiatkowski, F.; Chalabi, N.; Satih, S.; Bignon, Y.J.; Bernard-Gallon, D.J. Central adiposity as a major risk factor of ovarian cancer. Anticancer Res. 2009, 29, 5229–5234. [Google Scholar]
- Risch, H.A. Hormonal etiology of epithelial ovarian cancer, with a hypothesis concerning the role of androgens and progesterone. J. Natl. Cancer Inst. 1998, 90, 1774–1786. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef] [PubMed]
- Hoyo, C.; Berchuck, A.; Halabi, S.; Bentley, R.C.; Moorman, P.; Calingaert, B.; Schildkraut, J.M. Anthropometric measurements and epithelial ovarian cancer risk in African-American and White women. Cancer Causes Control 2005, 16, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Coletta, A.M.; Allen, P.K.; Parikh, A.M.; Cox-Mattin, M.; Meyer, L.A.; Sun, C.C.; Basen-Engquist, K.M.; Lu, K.H.; Klopp, A.H. Perirenal Adiposity is Associated with Lower Progression-Free Survival from Ovarian Cancer. Int. J. Gynecol. Cancer 2018, 28, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hiruma, T.; Koizumi, M.; Yoshihara, M.; Nakamura, Y.; Tadokoro, H.; Motomatsu, S.; Yamanaka, T.; Washimi, K.; Okubo, Y.; et al. Bone marrow adipocytes induce cancer-associated fibroblasts and immune evasion, enhancing invasion and drug resistance. Cancer Sci. 2023, 114, 2674–2688. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Lee, B.; Kim, M.K.; Gong, S.P.; Park, N.H.; Chung, H.H.; Kim, H.S.; No, J.H.; Park, W.Y.; Park, A.K.; et al. Gene expression profiles of human subcutaneous and visceral adipose-derived stem cells. Cell Biochem. Funct. 2016, 34, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, H.S.; Kim, S.; Haegeman, G.; Tsang, B.K.; Dhanasekaran, D.N.; Song, Y.S. Adipose Stromal Cells from Visceral and Subcutaneous Fat Facilitate Migration of Ovarian Cancer Cells via IL-6/JAK2/STAT3 Pathway. Cancer Res. Treat. 2017, 49, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, A.; Marini, F.C.; Solley, T.N.; Elizondo, P.B.; Zhang, Y.; Sharp, H.J.; Broaddus, R.; Kolonin, M.; Mok, S.C.; Thompson, M.S.; et al. Human omental-derived adipose stem cells increase ovarian cancer proliferation, migration, and chemoresistance. PLoS ONE 2013, 8, e81859. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes-bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Yao, H.; He, S. Multi-faceted role of cancer-associated adipocytes in the tumor microenvironment (Review). Mol. Med. Rep. 2021, 24, 866. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escuredo, J.; Van Hee, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef]
- Habanjar, O.; Diab-Assaf, M.; Caldefie-Chezet, F.; Delort, L. The Impact of Obesity, Adipose Tissue, and Tumor Microenvironment on Macrophage Polarization and Metastasis. Biology 2022, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Hilliard, T.S.; Wang, Z.; Johnson, J.; Wang, W.; Harper, E.I.; Ott, C.; O’Brien, C.; Campbell, L.; et al. Host obesity alters the ovarian tumor immune microenvironment and impacts response to standard of care chemotherapy. J. Exp. Clin. Cancer Res. 2023, 42, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Wang, L.; Cheng, M.; Yang, H. Fibroblast growth factor 21 is related to cisplatin resistance in ovarian cancer. Chin. Med. J. 2022, 135, 1500–1502. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Bandera, E.V.; Lee, V.S.; Qin, B.; Rodriguez-Rodriguez, L.; Powell, C.B.; Kushi, L.H. Impact of body mass index on ovarian cancer survival varies by stage. Br. J. Cancer 2017, 117, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Mittendorfer, B. Gender differences in lipid metabolism and the effect of obesity. Obstet. Gynecol. Clin. N. Am. 2009, 36, 245–265. [Google Scholar] [CrossRef]
- Demark-Wahnefried, W.; Schmitz, K.H.; Alfano, C.M.; Bail, J.R.; Goodwin, P.J.; Thomson, C.A.; Bradley, D.W.; Courneya, K.S.; Befort, C.A.; Denlinger, C.S.; et al. Weight management and physical activity throughout the cancer care continuum. CA Cancer J. Clin. 2018, 68, 64–89. [Google Scholar] [CrossRef]
- Novikoff, A.B. Electron microscopy: Cytology of cell fractions. Science 1956, 124, 969–972. [Google Scholar] [CrossRef]
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235. [Google Scholar] [CrossRef]
- Chakraborty, K.; Leung, K.; Krishnan, Y. High lumenal chloride in the lysosome is critical for lysosome function. eLife 2017, 6, e28862. [Google Scholar] [CrossRef]
- Futai, M.; Sun-Wada, G.-H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B 2019, 95, 261–277. [Google Scholar] [CrossRef]
- Pei, J.; Wang, G.; Feng, L.; Zhang, J.; Jiang, T.; Sun, Q.; Ouyang, L. Targeting Lysosomal Degradation Pathways: New Strategies and Techniques for Drug Discovery. J. Med. Chem. 2021, 64, 3493–3507. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Savini, M.; Zhao, Q.; Wang, M.C. Lysosomes: Signaling Hubs for Metabolic Sensing and Longevity. Trends Cell Biol. 2019, 29, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef] [PubMed]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 2019, 29, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Zoncu, R. The Lysosome at the Intersection of Cellular Growth and Destruction. Dev. Cell 2020, 54, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, K.; Long, A.; Jia, L.; Zhang, Y.; Deng, H.; Li, Y.; Han, J.; Wang, Y. Fasting-induced hormonal regulation of lysosomal function. Cell Res. 2017, 27, 748–763. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, S.; Khambu, B.; Ma, F.; Li, Y.; Chen, X.; Martina, J.A.; Puertollano, R.; Li, Y.; Chalasani, N.; et al. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy 2018, 14, 1779–1795. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.J.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hodge, J.; Liu, Q.; Wang, J.; Wang, Y.; Evans, T.D.; Altomare, D.; Yao, Y.; Murphy, E.A.; Razani, B.; et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J. Immunother. Cancer 2020, 8, e000543. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Kobayashi, M.; Tagawa, R.; Nakagawa, Y.; Shimano, H.; Higami, Y. Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 3688. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Hahn, Y.; Silverstein, B.; Singh, M.; Fleitz, A.; Van, J.; Chen, H.; Liang, Q. Lysosomal dysfunction in diabetic cardiomyopathy. Front. Aging 2023, 4, 1113200. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Osonoi, Y.; Mita, T.; Azuma, K.; Nakajima, K.; Masuyama, A.; Goto, H.; Nishida, Y.; Miyatsuka, T.; Fujitani, Y.; Koike, M.; et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 2018, 14, 1991–2006. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Volat, F.; Sandhow, L.; Galitzky, J.; Nguyen, T.; Esteve, D.; Astrom, G.; Mejhert, N.; Ledoux, S.; Thalamas, C.; et al. CD36 Is a Marker of Human Adipocyte Progenitors with Pronounced Adipogenic and Triglyceride Accumulation Potential. Stem Cells 2017, 35, 1799–1814. [Google Scholar] [CrossRef]
- Christiaens, V.; Van Hul, M.; Lijnen, H.R.; Scroyen, I. CD36 promotes adipocyte differentiation and adipogenesis. Biochim. Biophys. Acta 2012, 1820, 949–956. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells 2021, 10, 1833. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef]
- Woloszynek, J.C.; Coleman, T.; Semenkovich, C.F.; Sands, M.S. Lysosomal dysfunction results in altered energy balance. J. Biol. Chem. 2007, 282, 35765–35771. [Google Scholar] [CrossRef]
- Flaherty, S.E., 3rd; Grijalva, A.; Xu, X.; Ables, E.; Nomani, A.; Ferrante, A.W., Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019, 363, 989–993. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Tang, T.; Yang, Z.Y.; Wang, D.; Yang, X.Y.; Wang, J.; Li, L.; Wen, Q.; Gao, L.; Bian, X.W.; Yu, S.C. The role of lysosomes in cancer development and progression. Cell Biosci. 2020, 10, 131. [Google Scholar] [CrossRef]
- Li, T.F.; Zeng, H.J.; Shan, Z.; Ye, R.Y.; Cheang, T.Y.; Zhang, Y.J.; Lu, S.H.; Zhang, Q.; Shao, N.; Lin, Y. Overexpression of kinesin superfamily members as prognostic biomarkers of breast cancer. Cancer Cell Int. 2020, 20, 123. [Google Scholar] [CrossRef]
- Khaket, T.P.; Kwon, T.K.; Kang, S.C. Cathepsins: Potent regulators in carcinogenesis. Pharmacol. Ther. 2019, 198, 1–19. [Google Scholar] [CrossRef]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Alvarez, J.; Dilshat, R.; Fock, V.; Moller, K.; Karl, L.; Larue, L.; Ogmundsdottir, M.H.; Steingrimsson, E. MITF and TFEB cross-regulation in melanoma cells. PLoS ONE 2020, 15, e0238546. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Astanina, E.; Bussolino, F.; Doronzo, G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol. Oncol. 2021, 15, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Perera, R.M.; Kimmelman, A.C. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy 2020, 16, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Saftig, P. Physiology of the lysosome. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Joyce, H.; McCann, A.; Clynes, M.; Larkin, A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 795–809. [Google Scholar] [CrossRef]
- Halaby, R. Influence of lysosomal sequestration on multidrug resistance in cancer cells. Cancer Drug Resist. 2019, 2, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Muallem, S.; Kim, S.H.; Kwon, K.B.; Kim, M.S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys. Res. Commun. 2019, 510, 409–415. [Google Scholar] [CrossRef]
- Geisslinger, F.; Muller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance-A Mini Review. Front. Oncol. 2020, 10, 1156. [Google Scholar] [CrossRef]
- Noack, A.; Gericke, B.; von Kockritz-Blickwede, M.; Menze, A.; Noack, S.; Gerhauser, I.; Osten, F.; Naim, H.Y.; Loscher, W. Mechanism of drug extrusion by brain endothelial cells via lysosomal drug trapping and disposal by neutrophils. Proc. Natl. Acad. Sci. USA 2018, 115, E9590–E9599. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef]
- Goldman, S.D.; Funk, R.S.; Rajewski, R.A.; Krise, J.P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 2009, 1, 1445–1459. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Gillet, J.P.; Gottesman, M.M. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr. Pharm. Biotechnol. 2011, 12, 686–692. [Google Scholar] [CrossRef]
- Molinari, A.; Calcabrini, A.; Meschini, S.; Stringaro, A.; Crateri, P.; Toccacieli, L.; Marra, M.; Colone, M.; Cianfriglia, M.; Arancia, G. Subcellular detection and localization of the drug transporter P-glycoprotein in cultured tumor cells. Curr. Protein Pept. Sci. 2002, 3, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Al-Shumary, D.S.; Al-Shammari, A.M.; Rasheed, M.N. Increased Expression of the ABCA1 and ABCA3 Transporter Genes is Associated with Cisplatin Resistance in Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2023, 24, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, D.; Gillet, J.P.; Sauerbrey, A.; Gruhn, B.; Dawczynski, K.; Bertholet, V.; de Longueville, F.; Zintl, F.; Remacle, J.; Efferth, T. ABCA3 as a possible cause of drug resistance in childhood acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 4357–4363. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Graab, P.; Bock, C.; Weiss, K.; Hirth, A.; Koller, N.; Braner, M.; Jung, J.; Loehr, F.; Tampe, R.; Behrends, C.; et al. Lysosomal targeting of the ABC transporter TAPL is determined by membrane-localized charged residues. J. Biol. Chem. 2019, 294, 7308–7323. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.; Jung, J. Impact of Obesity and Lysosomal Dysfunction on Chemoresistance in Ovarian Cancer. Biomedicines 2024, 12, 604. https://doi.org/10.3390/biomedicines12030604
Kim B, Jung J. Impact of Obesity and Lysosomal Dysfunction on Chemoresistance in Ovarian Cancer. Biomedicines. 2024; 12(3):604. https://doi.org/10.3390/biomedicines12030604
Chicago/Turabian StyleKim, Boyun, and Jewon Jung. 2024. "Impact of Obesity and Lysosomal Dysfunction on Chemoresistance in Ovarian Cancer" Biomedicines 12, no. 3: 604. https://doi.org/10.3390/biomedicines12030604
APA StyleKim, B., & Jung, J. (2024). Impact of Obesity and Lysosomal Dysfunction on Chemoresistance in Ovarian Cancer. Biomedicines, 12(3), 604. https://doi.org/10.3390/biomedicines12030604