Role of HGF–MET Signaling in Primary and Acquired Resistance to Targeted Therapies in Cancer



Abstract
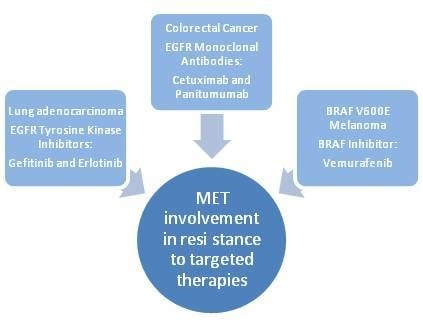
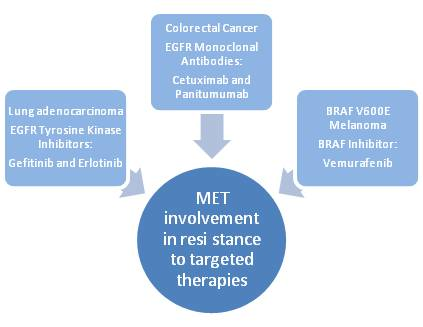
:
{kind=link}
1. Introduction
2. Role of MET in Mediating Resistance to Targeted Therapies in Human Cancers: The Examples of Lung Cancer, Colorectal Cancer and Melanoma
2.1. Lung Cancer
2.2. Colorectal Cancer
2.3. Melanoma
3. Research Gaps
Acknowledgments
Conflicts of Interest
References
- Aparicio, I.M.; Garcia-Marin, L.J.; Andreolotti, A.G.; Bodega, G.; Jensen, R.T.; Bragado, M.J. Hepatocyte growth factor activatesseveral transduction pathways in rat pancreatic acini. Biochim. Biophys. Acta 2003, 1643, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Nakayama, M.; Suzuki, Y.; Sakai, K.; Nakamura, T.; Sakai, Y.; Matsumoto, K. Suppression of acute hepatic injury by a synthetic prostacyclin agonist through hepatocyte growth factor expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G420–G429. [Google Scholar] [CrossRef]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Traut-wein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphoring receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.J.; El-Hefnawy, T.; Queiroz de Oliveira, P.E.; Raja, S.; Finkelstein, S.; Gooding, W.; Luketich, J.D.; Godfrey, T.E.; Hughes, S.J. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma. Neoplasia 2005, 7, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Bandla, S.; Pennathur, A.; Luketich, J.D.; Beer, D.G.; Lin, L.; Bass, A.J.; Godfrey, T.E.; Litle, V.R. Comparative genomics of esophageal adenocarcinoma and squamous cell carcinoma. Ann. Thorac. Surg. 2012, 93, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.L.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Rong, S.; Anver, M.; vande Woude, G.F. Autocrine hepatocyte growth factor/scatter factor-Met signaling induces transformation and the invasive/metastastic phenotype in C127 cells. Oncogene 1996, 13, 853–856. [Google Scholar] [PubMed]
- Rong, S.; Segal, S.; Anver, M.; Resau, J.H.; vande Woude, G.F. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrinestimulation. Proc. Natl. Acad. Sci. USA 1994, 91, 4731–4735. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, Y.L.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.J.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207. [Google Scholar] [PubMed]
- Daveau, M.; Scotte, M.; François, A.; Coulouarn, C.; Ros, G.; Tallet, Y.; Hiron, M.; Hellot, M.F.; Salier, J.P. Hepatocyte growth factor; transforming growth factor alpha; and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinog. 2003, 36, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; vande Woude, G.F.; et al. c-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome inde pendent of Her2/neu. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Bilchik, A.; Saha, S.; Turner, R.; Wiese, D.; Tanaka, M.; Kuo, C.; Wang, H.J.; Hoon, D.S. c-MET expression level in primary colon cancer: A predictor of tumor invasion and lymphnode metastases. Clin. Cancer Res. 2003, 9, 1480–1488. [Google Scholar] [PubMed]
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Romano, G.; di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.M.; Giordano, S.; Comoglio, P.M.; Ullrich, A. Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J. Biol. Chem. 2004, 279, 28970–28978. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin-like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther. 2006, 5, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.H.; Liu, H.E.; Lee, H.L.; Lin, C.L.; Chen, W.Y.; Wu, Z.H.; Lin, S.E.; Chiang, L.L.; Chung, C.L. Distinct clinical outcomes of non-small cell lung cancer patients with epidermal growth factor receptor (EGFR) mutations treated with EGFR tyrosine kinase inhibitors: Non-responders versus responders. PLoS One 2013, 8, e83266. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Caffarra, C.; Saccani, F.; Galvani, E.; Galetti, M.; Fumarola, C.; Bonelli, M.; Cavazzoni, A.; Cretella, D.; Sirangelo, R.; et al. Gefitinib inhibits invasive phenotype and epithelial-mesenchymal transition in drug-resistant NSCLC cells with MET amplification. PLoS One 2013, 8, e78656. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; von Pawel, J.; Garmey, E.G.; Akerley, W.L.; Brugger, W.; Ferrari, D.; Chen, Y.; Costa, D.B.; Gerber, D.E.; Orlov, S.; et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Novello, S.; Schiller, J.H.; Hirsh, V.; Sequist, L.V.; Soria, J.C.; von Pawel, J.; Schwartz, B.; von Roemeling, R.; Sandler, A.B. Rationale and design of MARQUEE: A phase III; randomized; double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic; nonsquamous; non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.; Daniel, D.B.; Goldschmidt, J.H., Jr.; Blumenschein, G.R., Jr.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Siegel, E.M.; Jacobsen, P.B.; Lee, J.H.; Malafa, M.; Fulp, W.; Fletcher, M.; Smith, J.C.; Brown, R.; Levine, R.; Cartwright, T.; et al. Florida Initiative for Quality Cancer Care: Improvements on colorectal cancer quality of care indicators during a 3-year interval. J. Am. Coll. Surg. 2014, 218, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Bertuccio, P.; Malvezzi, M.; Levi, F.; Chatenoud, L.; Negri, E.; la Vecchia, C. Cancer mortality in Europe; 2005–2009; and an overview of trends since 1980. Ann. Oncol. 2013, 24, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 27, 6550–6565. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Choi, J.S.; Seo, J.; Song, J.Y.; Lee, S.E.; Kwon, M.J.; Kwon, M.J.; Kundu, J.; Jung, K.; Oh, E.; et al. MET is a potential target for use in combination therapy with EGFR inhibition in triple-negative/basal-like breast cancer. Int. J. Cancer 2014, 134, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Annemans, L.; van Cutsem, E.; Humblet, Y.; van Laethem, J.L.; Bleiberg, H. Cost-effectiveness of cetuximab in combination with irinotecan compared with current care in metastatic colorectal cancer after failure on irinotecan—A Belgian analysis. Acta Clin. Belg. 2007, 62, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Wolf, M.; Peeters, M.; van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Sartore-Bianchi, A.; di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Jonker, D.J.; di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Bachet, J.B.; le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Douillard, J.Y.; Ducreux, M.; Peeters, M. Targeted therapy in metastatic colorectal cancer—An example of personalised medicine in action. Cancer Treat. Rev. 2013, 39, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; di Nicolantonio, F.; Nichelatti, M.; Molinari, F.; de Dosso, S.; Saletti, P.; Martini, M.; Cipani, T.; Marrapese, G.; Mazzucchelli, L.; et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One 2009, 4, e7287. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. Molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Normanno, N. HER2 signaling and resistance to the anti-EGFR monoclonal antibody cetuximab: A further step toward personalized medicine for patients with colorectal cancer. Cancer Discov. 2011, 1, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C.; et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Bachleitner-Hofmann, T.; Sun, M.Y.; Chen, C.T.; Liska, D.; Zeng, Z.; Viale, A.; Olshen, A.B.; Mittlboeck, M.; Christensen, J.G.; Rosen, N.; et al. Antitumor activity of SNX-2112; a synthetic heat shock protein-90 inhibitor; in MET-amplified tumor cells with or without resistance to selective MET Inhibition. Clin. Cancer Res. 2011, 17, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR–MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Tabernero, J.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.P.; Davidenko, I.; Chen, L.; Smethurst, D.; van Cutsem, E. Panitumumab (pmab) plus AMG 102 in patients (pts) with wild-type KRAS metastatic colorectal cancer (mCRC): Updated safety results. J. Clin. Oncol. 2010, 28, e14083. [Google Scholar]
- Van Cutsem, E.; Eng, C.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.; Davidenko, I.; Stephenson, J.; Elez, E.; Prenen, H.; et al. A randomized; phase Ib/II trial of rilotumumab (AMG 102, ril) or ganitumumab (AMG 479, gan) with panitumumab (pmab) versus pmab alone in patients (pts) with wild-type (WT) KRAS metastatic colorectal (mCRC): Primary and biomarker analyses. J. Clin. Oncol. 2011, 29, 3500. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Ervin, T.J.; Gallinson, D.; Singh, J.; Wallace, J.A.; Saleh, M.N.; Vallone, M.; Phan, S.C.; Hack, S.P. Treatment rationale and study design for a randomized; double-blind; placebo-controlled phase II study evaluating onartuzumab (MetMAb) in combination with bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal cancer. Clin. Colorectal Cancer 2013, 12, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Bessudo, A.; Bendell, J.C.; Gabrail, N.; Kopp, M.V.; Mueller, L.; Hart, L.L.; Vladimirov, V.I.; Pande, A.U.; Gorbatchevsky, I.; Eng, C. Phase I results of the randomized; placebo controlled; phase I/II study of the novel oral c-Met inhibitor, ARQ 197, irinotecan (cpt-11), and cetuximab (C) in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC) who received front-line systemic therapy. J. Clin. Oncol. 2011, 29, 527. [Google Scholar] [CrossRef]
- Eng, C.; Hart, L.L.; Severtsev, A.; Gladkov, O.; Mueller, L.; Kopp, M.V.; Vladimirov, V.I.; Langdon, R.M.; Kotiv, B.; Barni, S. A randomized; placebo-controlled; phase I/II study of tivantinib (ARQ 197) in combination with cetuximab and irinotecan in patients (pts) with KRAS wild-type (WT) metastatic colorectal cancer (CRC) who had received previous front-line systemic therapy. J. Clin. Oncol. 2013, 31. abstr. 3508. [Google Scholar]
- Hodi, F.S.; OʼDay, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; OʼDay, S.J.W.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; Davidson, N.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shah, S.R.; Illum, H.; Dowell, J. Vemurafenib: Targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 2012, 72, 2207–2022. [Google Scholar] [CrossRef] [PubMed]
- Natali, P.G.; Nicotra, M.R.; di Renzo, M.F.; Prat, M.; Bigotti, A.; Cavaliere, R.; Comoglio, P.M. Expression of the c-Met/HGF receptor in human melanocytic neoplasms: Demonstration of the relationship to malignant melanoma tumour progression. Br. J. Cancer 1993, 68, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T. Resistance patterns with tyrosine kinase inhibitors in melanoma: New insights. Curr. Opin. Oncol. 2012, 24, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Ahmed, S.; Janamanchi, V.; Tretiakova, M.; Zumba, O.; Krausz, T.; Jagadeeswaran, R.; Salgia, R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin. Cancer Res. 2007, 13, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Vergani, E.; Vallacchi, V.; Frigerio, S.; Deho, P.; Mondellini, P.; Perego, P.; Cassinelli, G.; Lanzi, C.; Testi, M.A.; Rivoltini, L.; et al. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia 2011, 13, 1132–1142. [Google Scholar] [PubMed]
- Fufa, T.D.; Pavan, W.J. Axing the cancer loop. Pigment. Cell Melanoma Res. 2014, 27, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, D.U.; Lee, D.; Reed, X.; Fletez-Brant, C.; Bessling, S.L.; Loftus, S.K.; Beer, M.A.; Pavan, W.J.; Mccallion, A.S. Integration of Chip-Seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res. 2012, 22, 2290–2301. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.E.; Barajas, B.; Bussat, R.T.; Yan, K.J.; Neela, P.H.; Flockhart, R.J.; Kovalski, J.; Zehnder, A.; Khavari, P.A. Enhancer-targeted genome editing selectively blocks innate resistance to oncokinase inhibition. Genome Res. 2014, 24, 751–760. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Corte, C.M.; Fasano, M.; Papaccio, F.; Ciardiello, F.; Morgillo, F. Role of HGF–MET Signaling in Primary and Acquired Resistance to Targeted Therapies in Cancer. Biomedicines 2014, 2, 345-358. https://doi.org/10.3390/biomedicines2040345
Della Corte CM, Fasano M, Papaccio F, Ciardiello F, Morgillo F. Role of HGF–MET Signaling in Primary and Acquired Resistance to Targeted Therapies in Cancer. Biomedicines. 2014; 2(4):345-358. https://doi.org/10.3390/biomedicines2040345
Chicago/Turabian StyleDella Corte, Carminia Maria, Morena Fasano, Federica Papaccio, Fortunato Ciardiello, and Floriana Morgillo. 2014. "Role of HGF–MET Signaling in Primary and Acquired Resistance to Targeted Therapies in Cancer" Biomedicines 2, no. 4: 345-358. https://doi.org/10.3390/biomedicines2040345
APA StyleDella Corte, C. M., Fasano, M., Papaccio, F., Ciardiello, F., & Morgillo, F. (2014). Role of HGF–MET Signaling in Primary and Acquired Resistance to Targeted Therapies in Cancer. Biomedicines, 2(4), 345-358. https://doi.org/10.3390/biomedicines2040345