Hosts for Hostile Protein Production: The Challenge of Recombinant Immunotoxin Expression



{kind=link}
{kind=link}
Abstract
:1. Introduction
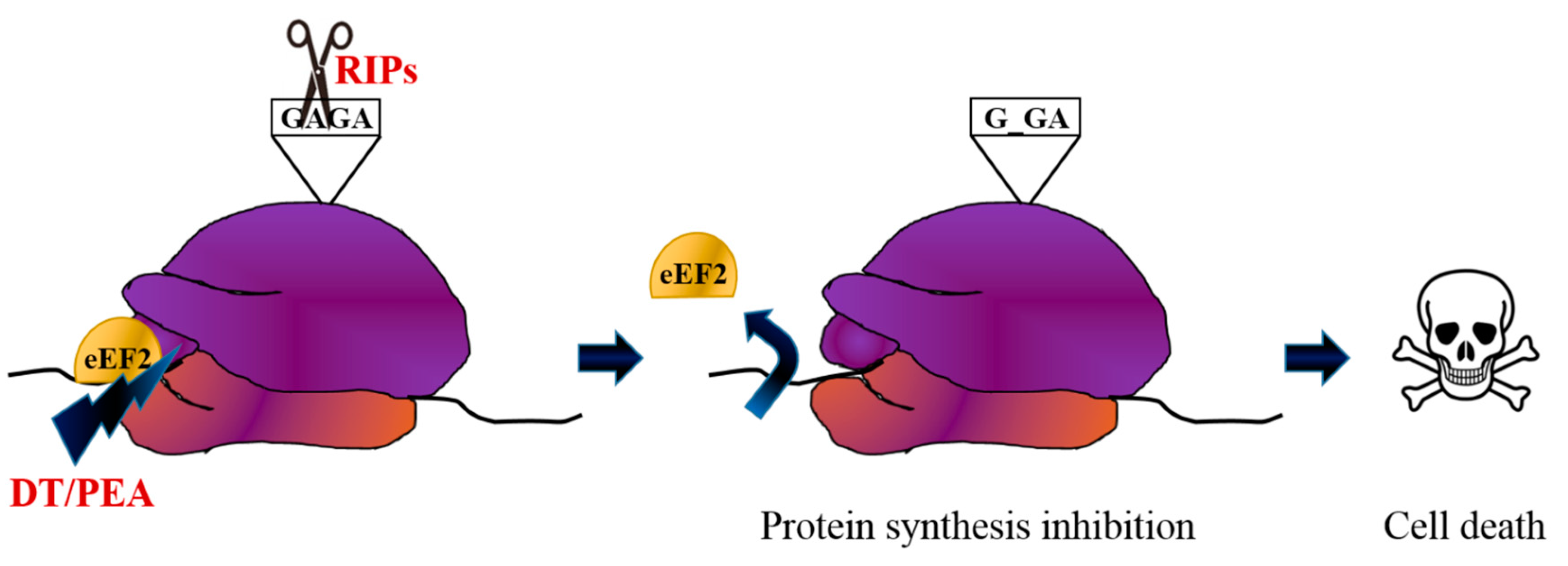
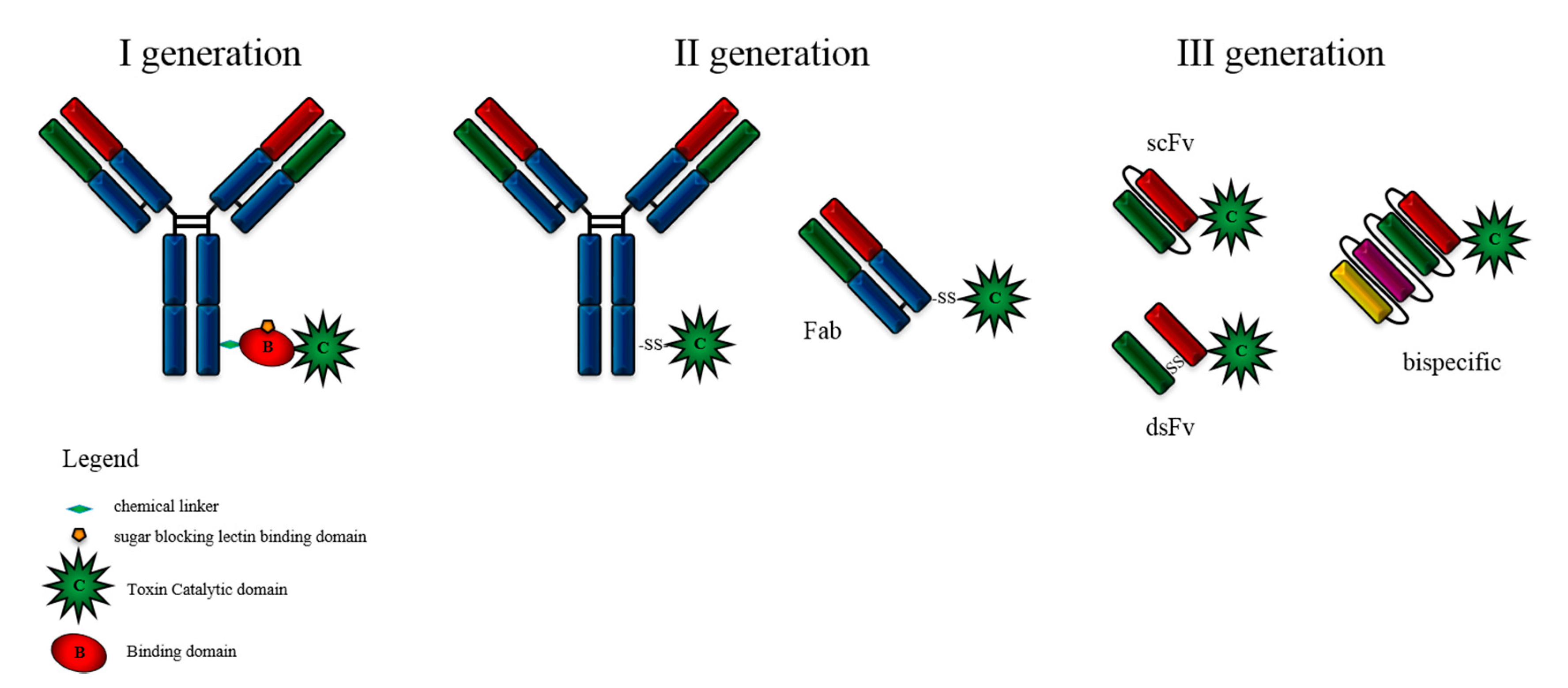
2. Immunotoxin Development
3. Immunotoxin Factories
3.1. Bacteria
3.2. Intracellular Immunization and Issues Related to Host Auto-Intoxication
3.3. Yeast
3.4. Plant Cells
3.5. Toxin-Resistant Cells
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar] [CrossRef]
- Williams, D.P.; Snider, C.E.; Strom, T.B.; Murphy, J.R. Structure/function analysis of interleukin-2-toxin (DAB486-IL-2). Fragment B sequences required for the delivery of fragment A to the cytosol of target cells. J. Biol. Chem. 1990, 265, 11885–11889. [Google Scholar] [PubMed]
- Cheung, L.S.; Fu, J.; Kumar, P.; Kumar, A.; Urbanowski, M.E.; Ihms, E.A.; Parveen, S.; Bullen, C.K.; Patrick, G.J.; Harrison, R.; et al. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3100–3105. [Google Scholar] [CrossRef]
- Madhumathi, J.; Devilakshmi, S.; Sridevi, S.; Verma, R.S. Immunotoxin therapy for hematologic malignancies: Where are we heading? Drug Discov. Today 2016, 21, 325–332. [Google Scholar] [CrossRef]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, D.J. Countering immunotoxin immunogenicity. Br. J. Cancer 2016, 114, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhar, I.; Padlan, E.A.; Jung, S.H.; Lee, B.; Pastan, I. Rapid humanization of the Fv of monoclonal antibody B3 by using framework exchange of the recombinant immunotoxin B3(Fv)-PE38. Proc. Natl. Acad. Sci. USA 1994, 91, 12051–12055. [Google Scholar] [CrossRef]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.-C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, F.; Flavell, D.J.; Angelucci, F.; Fabbrini, M.S.; Ippoliti, R. Strategies to Improve the Clinical Utility of Saporin-Based Targeted Toxins. Toxins 2018, 10, 82. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Todhunter, D.; Oh, S.; Vallera, D.A.; Choi, S.H. Mutagenic Deimmunization of Diphtheria Toxin for Use in Biologic Drug Development. Toxins 2015, 7, 4067–4082. [Google Scholar] [CrossRef] [Green Version]
- Bera, T.K.; Onda, M.; Kreitman, R.J.; Pastan, I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk. Res. 2014, 38, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Cizeau, J.; Grenkow, D.M.; Brown, J.G.; Entwistle, J.; Macdonald, G.C. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Wu, Y.; Yan, J.-Y.; Gao, Y.; Wang, Y.; Mi, S.-L.; An, C.-C. Y55 and D78 are crucial amino acid residues of a new IgE epitope on trichosanthin. Biochem. Biophys. Res. Commun. 2006, 343, 1251–1256. [Google Scholar] [CrossRef]
- Leung, K.C.; Meng, Z.Q.; Ho, W.K. Antigenic determination fragments of alpha-momorcharin. Biochim. Biophys. Acta 1997, 1336, 419–424. [Google Scholar] [CrossRef]
- Smallshaw, J.E.; Ghetie, V.; Rizo, J.; Fulmer, J.R.; Trahan, L.L.; Ghetie, M.-A.; Vitetta, E.S. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat. Biotechnol. 2003, 21, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dai, J.; Li, B.; Fan, K.; Peng, L.; Zhang, D.; Cao, Z.; Qian, W.; Wang, H.; Zhao, J.; et al. Expression, purification and characterization of an immunotoxin containing a humanized anti-CD25 single-chain fragment variable antibody fused to a modified truncated Pseudomonas exotoxin A. Protein Expr. Purif. 2008, 58, 140–147. [Google Scholar] [CrossRef]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Fabbrini, M.S.; Katayama, M.; Nakase, I.; Vago, R.; Barbier, J.; Gillet, D. Plant ribosome-inactivating proteins: Progesses, challenges and biotechnological applications (and a few digressions). Toxins (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Vago, R.; Ippoliti, R.; Fabbrini, M.S. Current status & Biomedical applications of Ribosome-inactivating proteins. In Antitumor Potential and other Emerging Medicinal Properties of Natural Compounds; Fang, E., Ng, T., Eds.; Springer: Berlin, Germany, 2013; pp. 145–179. [Google Scholar]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef]
- Rust, A.; Partridge, L.J.; Davletov, B.; Hautbergue, G.M.; Barbier, J.; Gillet, D. The use of plant-derived ribosome inactivating proteins in immunotoxin development: Past, present and future generations. Toxins (Basel) 2017, 9, 344. [Google Scholar] [CrossRef]
- Itakura, K.; Hirose, T.; Crea, R.; Riggs, A.; Heyneker, H.; Bolivar, F.; Boyer, H. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 1977, 198, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, A.A.; Viitanen, P.V.; Lorimer, G.H. Chaperonin assisted polypeptide folding and assembly: Implications for the production of functional proteins in bacteria. Trends Biotechnol. 1990, 8, 354–358. [Google Scholar] [CrossRef]
- Dullah, E.C.; Ongkudon, C.M. Current trends in endotoxin detection and analysis of endotoxin-protein interactions. Crit. Rev. Biotechnol. 2017, 37, 251–261. [Google Scholar] [CrossRef]
- Smith, G.E.; Summers, M.D.; Fraser, M.J. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell Biol. 1983, 3, 2156–2165. [Google Scholar] [CrossRef]
- Cregg, J.M.; Vedvick, T.S.; Raschke, W.C. Recent advances in the expression of foreign genes in Pichia pastoris. Biotechnology 1993, 11, 905–910. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Higel, F.; Seidl, A.; Sörgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Taunt, H.N.; Stoffels, L.; Purton, S. Green biologics: The algal chloroplast as a platform for making biopharmaceuticals. Bioengineered 2018, 9, 48–54. [Google Scholar] [CrossRef]
- Vallera, D.A.; Oh, S.; Chen, H.; Shu, Y.; Frankel, A.E. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human CD22 and CD19 receptors in a mouse model of B-cell metastases. Mol. Cancer Ther. 2010, 9, 1872–1883. [Google Scholar] [CrossRef]
- Della Cristina, P.; Castagna, M.; Lombardi, A.; Barison, E.; Tagliabue, G.; Ceriotti, A.; Koutris, I.; Di Leandro, L.; Giansanti, F.; Vago, R.; et al. Systematic comparison of single-chain Fv antibody-fusion toxin constructs containing Pseudomonas Exotoxin A or saporin produced in different microbial expression systems. Microb. Cell Fact. 2015, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.R.; Legname, G.; Osborn, R.; Chen, Z.; Lord, J. Single-chain ribosome inactivating proteins from plants depurinate Escherichia coli 23S ribosomal RNA. FEBS Lett. 1991, 290, 65–68. [Google Scholar] [CrossRef]
- Barthelemy, I.; Martineau, D.; Ong, M.; Matsunami, R.; Ling, N.; Benatti, L.; Cavallaro, U.; Soria, M.; A Lappi, D. The expression of saporin, a ribosome-inactivating protein from the plant Saponaria officinalis, in Escherichia coli. J. Biol. Chem. 1993, 268, 6541–6548. [Google Scholar] [PubMed]
- Chaddock, J.A.; Lord, J.; Hartley, M.R.; Roberts, L.M. Pokeweed antiviral protein (PAP) mutations which permit E. coli growth do not eliminate catalytic activity towards prokaryotic ribosomes. Nucleic Acids Res. 1994, 22, 1536–1540. [Google Scholar]
- Vago, R.; Marsden, C.J.; Lord, J.M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.-U.; Ceriotti, A.; Fabbrini, M.S. Saporin and ricin A chain follow different intracellular routes to enter the cytosol of intoxicated cells. FEBS J. 2005, 272, 4983–4995. [Google Scholar] [CrossRef] [Green Version]
- Joshi, B.H.; Puri, R.K. Optimization of expression and purification of two biologically active chimeric fusion proteins that consist of human interleukin-13 and Pseudomonas exotoxin in Escherichia coli. Protein Expr. Purif. 2005, 39, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.K.; Queen, G.; Junghans, R.P.; Waldmann, T.A.; Fitzgerald, D.J.; Pastan, I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 1989, 339, 394–397. [Google Scholar] [CrossRef]
- Brinkmann, U.; Reiter, Y.; Jung, S.H.; Lee, B.; Pastan, I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc. Natl. Acad. Sci. USA 1993, 90, 7538–7542. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zhang, Z.; Shen, X.; Zhong, F. Codon optimization enhances secretory expression of Pseudomonas aeruginosa exotoxin A in E. coli. Protein Expr. Purif. 2010, 72, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, V.; Bao, X.; Keir, S.T.; Pegram, C.N.; Szafranski, S.E.; Piao, H.; Wikstrand, C.J.; McLendon, R.E.; Kuan, C.-T.; Pastan, I.H.; et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin. Cancer Res. 2013, 19, 4717–4727. [Google Scholar] [CrossRef] [Green Version]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar] [CrossRef]
- Salvatore, G.; Beers, R.; Margulies, I.; Kreitman, R.J.; Pastan, I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res. 2002, 8, 995–1002. [Google Scholar]
- Alderson, R.F.; Kreitman, R.J.; Chen, T.; Yeung, P.; Herbst, R.; Fox, J.A.; Pastan, I. CAT-8015: A second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin. Cancer Res. 2009, 15, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutré, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Santiago, L.; Gao, G.; Lanasa, M.C.; et al. Minimal residual hairy cell leukemia eradication with moxetumomab pasudotox: Phase 1 results and long-term follow-up. Blood 2018, 131, 2331–2334. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Immunoconjugates in the management of hairy cell leukemia. Best Pract. Res. Clin. Haematol. 2015, 28, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Ramage, J.; Kiser, M.; Alexander, R.; Kucera, G.; Miller, M.S. Characterization of diphtheria fusion proteins targeted to the human interleukin-3 receptor. Protein Eng. 2000, 13, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.; Liu, J.-S.; Rizzieri, D.; Hogge, D. Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk. Lymphoma 2008, 49, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Woo, J.H.; Ahn, C.; Pemmaraju, N.; Medeiros, B.C.; Carraway, H.E.; Frankfurt, O.; Forman, S.J.; Yang, X.A.; Konopleva, M.; et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood 2014, 124, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef]
- Marshall, R.S.; D’Avila, F.; Di Cola, A.; Traini, R.; Spanò, L.; Fabbrini, M.S.; Ceriotti, A. Signal peptide-regulated toxicity of a plant ribosome-inactivating protein during cell stress. Plant J. 2011, 65, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, M.S.; Carpani, D.; Soria, M.R.; Ceriotti, A. Cytosolic immunization allows the expression of preATF-saporin chimeric toxin in eukaryotic cells. FASEB J. 2000, 14, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Boldicke, T. Single domain antibodies for the knockdown of cytosolic and nuclear proteins. Protein Sci. 2017, 26, 925–945. [Google Scholar] [CrossRef] [Green Version]
- Bragonzi, A.; Distefano, G.; Buckberry, L.D.; Acerbis, G.; Foglieni, C.; LaMotte, D.; Campi, G.; Marc, A.; Soria, M.R.; Jenkins, N.; et al. A new Chinese hamster ovary cell line expressing alpha2,6-sialyltransferase used as universal host for the production of human-like sialylated recombinant glycoproteins. Biochim. Biophys. Acta 2000, 1474, 273–282. [Google Scholar] [CrossRef]
- Tejwani, V.; Andersen, M.R.; Nam, J.H.; Sharfstein, S.T. Glycoengineering in CHO cells: Advances in systems biology. Biotechnol. J. 2018, 13, e1700234. [Google Scholar] [CrossRef] [PubMed]
- Rajamohan, F.; Doumbia, S.O.; Engstrom, C.R.; Pendergras, S.L.; Maher, D.L.; Uckun, F.M. Expression of biologically active recombinant pokeweed antiviral protein in methylotrophic yeast Pichia pastoris. Protein Expr. Purif. 2000, 18, 193–201. [Google Scholar] [CrossRef]
- Woo, J.H.; Liu, Y.Y.; Mathias, A.; Stavrou, S.; Wang, Z.; Thompson, J.; Neville, D.M. Gene optimization is necessary to express a bivalent anti-human anti-T cell immunotoxin in Pichia pastoris. Protein Expr. Purif. 2002, 25, 270–282. [Google Scholar] [CrossRef]
- Lombardi, A.; Bursomanno, S.; Lopardo, T.; Traini, R.; Colombatti, M.; Ippoliti, R.; Flavell, D.J.; Ceriotti, A.; Fabbrini, M.S. Pichia pastoris as a host for secretion of toxic saporin chimeras. FASEB J. 2010, 24, 253–265. [Google Scholar] [CrossRef]
- Provenzano, A.E.; Posteri, R.; Giansanti, F.; Angelucci, F.; Flavell, S.U.; Flavell, D.J.; Fabbrini, M.S.; Porro, D.; Ippoliti, R.; Ceriotti, A.; et al. Optimization of construct design and fermentation strategy for the production of bioactive ATF-SAP, a saporin based anti-tumoral uPAR-targeted chimera. Microb. Cell Fact. 2016, 15, 194. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Chang, W.; Zhang, K.; Cui, M.; Wu, S.; Xu, T. Expression and purification of recombinant ATF-mellitin, a new type fusion protein targeting ovarian cancer cells, in P. pastoris. Oncol. Rep. 2016, 35, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Du, Q.; Sturm, M.B.; Schramm, V.L. Soapwort saporin L3 expression in yeast, mutagenesis and RNA substrate specificity. Biochemistry 2015, 54, 4565–4574. [Google Scholar] [CrossRef]
- Wang, Z.; Louras, N.J.; Lellouch, A.G.; Pratts, S.G.; Zhang, H.; Wang, H.; Huang, C.A.; Cetrulo, C.L., Jr.; Madsen, J.C.; Sachs, D.H.; et al. Dosing optimization of CCR4 immunotoxin for improved depletion of CCR4(+) Treg in nonhuman primates. Mol. Oncol. 2018, 12, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Idiris, A.; Tohda, H.; Sasaki, M.; Okada, K.; Kumagai, H.; Giga-Hama, Y.; Takegawa, K. Enhanced protein secretion from multiprotease-deficient fission yeast by modification of its vacuolar protein sorting pathway. Appl. Microbiol. Biotechnol. 2010, 85, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef]
- Frigerio, L.; Boone, A.N.; Chan, A.; Kulpa, J.E.; Brownsey, R.W.; Vitale, A.; Lord, J.M.; Ceriotti, A.; Roberts, L.M. Free ricin A chain, proricin and native toxin have different cellular fates when expressed in tobacco protoplasts. J. Biol. Chem. 1998, 273, 14194–14199. [Google Scholar] [CrossRef]
- Krishnan, R.; McDonald, K.A.; Dandekar, A.M.; Jackman, A.P.; Falk, B. Expression of recombinant trichosanthin, a ribosome-inactivating protein, in transgenic tobacco. J. Biotechnol. 2002, 97, 69–88. [Google Scholar] [CrossRef]
- Dyo, Y.M.; Purton, S. The algal chloroplast as a synthetic biology platform for production of therapeutic proteins. Microbiology 2018, 164, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Surzycki, R.; Greenham, K.; Kitayama, K.; Dibal, F.; Wagner, R.; Rochaix, J.-D.; Ajam, T.; Surzycki, S.; Greenham, K. Factors effecting expression of vaccines in microalgae. Biologicals 2009, 37, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.R. Recombinant expression systems in the pharmaceutical industry. Appl. Microbiol. Biotechnol. 2004, 65, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Hempel, F.; Lau, J.; Klingl, A.; Maier, U.G. Algae as protein factories: Expression of a human antibody and the respective antigen in the diatom Phaeodactylum tricornutum. PLoS ONE 2011, 6, e28424. [Google Scholar] [CrossRef]
- Hempel, F.; Maier, U.G. An engineered diatom acting like a plasma cell secreting human IgG antibodies with high efficiency. Microb. Cell Fact. 2012, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Boynton, J.; Gillham, N.; Harris, E.; Hosler, J.; Johnson, A.; Jones, A.; Randolph-Anderson, B.; Robertson, D.; Klein, T.; Shark, K.; et al. Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 1988, 240, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Conrad, U.; Fiedler, U. Compartment-specific accumulation of recombinant immunoglobulins in plant cells: An essential tool for antibody production and immunomodulation of physiological functions and pathogen activity. Plant Mol. Biol. 1998, 38, 101–109. [Google Scholar] [CrossRef]
- Ma, J.K.; Drake, P.M.; Christou, P. The production of recombinant pharmaceutical proteins in plants. Nat. Rev. Genet. 2003, 4, 794–805. [Google Scholar] [CrossRef]
- Jarvis, P. Targeting of nucleus-encoded proteins to chloroplasts in plants. New Phytol. 2008, 179, 257–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dove, A. Uncorking the biomanufacturing bottleneck. Nat. Biotechnol. 2002, 20, 777–779. [Google Scholar] [CrossRef]
- Tran, M.; Zhou, B.; Pettersson, P.L.; Gonzalez, M.J.; Mayfield, S.P. Synthesis and assembly of a full-length human monoclonal antibody in algal chloroplasts. Biotechnol. Bioeng. 2009, 104, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.M.; Boynton, J.E.; Gillham, N.W.; Randolph-Anderson, B.L.; Johnson, A.M.; Harris, E.H. Transformation of chloroplast ribosomal RNA genes in Chlamydomonas: Molecular and genetic characterization of integration events. Genetics 1990, 126, 875–888. [Google Scholar]
- Goldschmidt-Clermont, M. Transgenic expression of aminoglycoside adenine transferase in the chloroplast: A selectable marker of site-directed transformation of chlamydomonas. Nucleic Acids Res. 1991, 19, 4083–4089. [Google Scholar] [CrossRef]
- Marín-Navarro, J.; Manuell, A.L.; Wu, J.; Mayfield, S.P. Chloroplast translation regulation. Photosynth. Res. 2007, 94, 359–374. [Google Scholar] [CrossRef]
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 2010, 61, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Choquet, Y.; Stern, D.B.; Wostrikoff, K.; Kuras, R.; Girard-Bascou, J.; Wollman, F.-A. Translation of cytochrome f is autoregulated through the 5’ untranslated region of petA mRNA in Chlamydomonas chloroplasts. Proc. Natl. Acad. Sci. USA 1998, 95, 4380–4385. [Google Scholar] [CrossRef]
- Ishikura, K.; Takaoka, Y.; Kato, K.; Sekine, M.; Yoshida, K.; Shinmyo, A. Expression of a foreign gene in Chlamydomonas reinhardtii chloroplast. J. Biosci. Bioeng. 1999, 87, 307–314. [Google Scholar] [CrossRef]
- Kasai, S.; Yoshimura, S.; Ishikura, K.; Takaoka, Y.; Kobayashi, K.; Kato, K.; Shinmyo, A. Effect of coding regions on chloroplast gene expression in Chlamydomonas reinhardtii. J. Biosci. Bioeng. 2003, 95, 276–282. [Google Scholar] [CrossRef]
- Minai, L.; Wostrikoff, K.; Wollman, F.-A.; Choquet, Y. Chloroplast biogenesis of photosystem II cores involves a series of assembly-controlled steps that regulate translation. Plant Cell 2006, 18, 159–175. [Google Scholar] [CrossRef]
- Manuell, A.L.; Quispe, J.; Mayfield, S.P. Structure of the chloroplast ribosome: Novel domains for translation regulation. PLoS Biol. 2007, 5, e209. [Google Scholar] [CrossRef]
- Michelet, L.; Lefebvre-Legendre, L.; Burr, S.E.; Rochaix, J.D.; Goldschmidt-Clermont, M. Enhanced chloroplast transgene expression in a nuclear mutant of Chlamydomonas. Plant Biotechnol. J. 2011, 9, 565–574. [Google Scholar] [CrossRef]
- Gallie, D.R. A tale of two termini: A functional interaction between the termini of an mRNA is a prerequisite for efficient translation initiation. Gene 1998, 216, 1–11. [Google Scholar] [CrossRef]
- Wells, S.E.; Hillner, P.E.; Vale, R.D.; Sachs, A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 1998, 2, 135–140. [Google Scholar] [CrossRef]
- Eberhard, S.; Drapier, D.; Wollman, F.A. Searching limiting steps in the expression of chloroplast-encoded proteins: Relations between gene copy number, transcription, transcript abundance and translation rate in the chloroplast of Chlamydomonas reinhardtii. Plant J. 2002, 31, 149–160. [Google Scholar] [CrossRef]
- Nickelsen, J. Chloroplast RNA-binding proteins. Curr. Genet. 2003, 43, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, D.; Franklin, S.; Schultz, J.; Henry, R.; Brown, E.; Coragliotti, A.; Mayfield, S.P. Contribution of 5′- and 3′-untranslated regions of plastid mRNAs to the expression of Chlamydomonas reinhardtii chloroplast genes. Mol. Genet. Genom. 2005, 274, 625–636. [Google Scholar] [CrossRef]
- Rasala, B.A.; Muto, M.; Lee, P.A.; Jager, M.; Cardoso, R.M.; Behnke, C.A.; Kirk, P.; Hokanson, C.A.; Crea, R.; Mendez, M.; et al. Production of therapeutic proteins in algae, analysis of expression of seven human proteins in the chloroplast of Chlamydomonas reinhardtii. Plant Biotechnol. J. 2010, 8, 719–733. [Google Scholar] [CrossRef]
- Mayfield, S.P.; Franklin, S.E.; Lerner, R.A. Expression and assembly of a fully active antibody in algae. Proc. Natl. Acad. Sci. USA 2003, 100, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, M.; Henry, R.E.; Siefker, D.; Van, C.; Newkirk, G.; Kim, J.; Bui, J.; Mayfield, S.P. Production of anti-cancer immunotoxins in algae: Ribosome inactivating proteins as fusion partners. Biotechnol. Bioeng. 2013, 110, 2826–2835. [Google Scholar] [CrossRef]
- Tran, M.; Van, C.; Barrera, D.J.; Pettersson, P.L.; Peinado, C.D.; Bui, J.; Mayfield, S.P. Production of unique immunotoxin cancer therapeutics in algal chloroplasts. Proc. Natl. Acad. Sci. USA 2013, 110, E15–E22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Woo, J.H.; Neville, D.M. Targeted introduction of a diphtheria toxin resistant mutation into the chromosomal EF-2 locus of Pichia pastoris and expression of immunotoxin in the EF-2 mutants. Protein Expr. Purif. 2003, 30, 262–274. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Gordienko, I.; Mathias, A.; Ma, S.; Thompson, J.; Woo, J.H.; Neville, D.M. Expression of an anti-CD3 single-chain immunotoxin with a truncated diphtheria toxin in a mutant CHO cell line. Protein Expr. Purif. 2000, 19, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Stavrou, S.; Weetall, M.; Hexham, J.; Digan, M.E.; Wang, Z.; Woo, J.H.; Yu, Y.; Mathias, A.; Liu, Y.Y.; et al. Improved binding of a bivalent single-chain immunotoxin results in increased efficacy for in vivo T-cell depletion. Protein Eng. 2001, 14, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, N.; Parekh, R.B.; James, D.C. Getting the glycosylation right: Implications for the biotechnology industry. Nat. Biotechnol. 1996, 14, 975–981. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuppone, S.; Fabbrini, M.S.; Vago, R. Hosts for Hostile Protein Production: The Challenge of Recombinant Immunotoxin Expression. Biomedicines 2019, 7, 38. https://doi.org/10.3390/biomedicines7020038
Zuppone S, Fabbrini MS, Vago R. Hosts for Hostile Protein Production: The Challenge of Recombinant Immunotoxin Expression. Biomedicines. 2019; 7(2):38. https://doi.org/10.3390/biomedicines7020038
Chicago/Turabian StyleZuppone, Stefania, Maria Serena Fabbrini, and Riccardo Vago. 2019. "Hosts for Hostile Protein Production: The Challenge of Recombinant Immunotoxin Expression" Biomedicines 7, no. 2: 38. https://doi.org/10.3390/biomedicines7020038
APA StyleZuppone, S., Fabbrini, M. S., & Vago, R. (2019). Hosts for Hostile Protein Production: The Challenge of Recombinant Immunotoxin Expression. Biomedicines, 7(2), 38. https://doi.org/10.3390/biomedicines7020038