Novel Pathogenic Variants of the AIRE Gene in Two Autoimmune Polyendocrine Syndrome Type I Cases with Atypical Presentation: Role of the NGS in Diagnostic Pathway and Review of the Literature



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients.
2.2. Sample Preparation and Next Generation Sequencing Analysis
2.3. Bioinformatic Pipeline
2.4. Multialignment of AIRE Orthologous Sequences
3. Results
3.1. NGS
3.2. ClustalW Analysis
4. Discussion
- Mucosa infection/inflammation: chronic MC;
- Impaired endocrine organ functions: CM, AD, hypothyroidism, panhypopituitarism;
- Ectodermal dystrophy (nail distrophy, enamel hypoplasia, alopecia, vitiligo, keratopathy).
- Chronic Mucocutaneous Candidiadis: it is present in up to 80% of all the patients aged one to three years and is mostly located at the oral mucosa and esophagus while, less frequently, it affects vaginal or intestinal and nail mucosa. Chronic infection makes the mucosa susceptible to squamous cell carcinoma of the mouth or of the esophagus in 5% of cases [17,18];
- Chronic Hypoparathyroidism is present in up to 80–90% of patients aged 10–15 years and characterized by paresthesias and tetany (mimicking an epileptic seizure) with hypocalcemia, hypophosphoremia, and a low PTH level. Autoantibodies NALP5 against parathyroids glands can be present in 11–38% of patients [19];
- Addison disease is the third late sign to appear, at around 13–14 years of age, and it consists in the nearly complete atrophy of the adrenal gland. It manifests with asthenia, hypotension, weight loss, hyperpigmentation of the skin, and mucosa [20]. The main biochemical feature is the lack of cortisol secretion after the ACTH stimulation test. This is due to the presence of autoantibodies against the adrenal cortex (ACA) and the 21-OH hydroxylasis enzyme. These antibodies are specific of the disease being present in up to 50% of patients with MC and CH [21].
- -
- Pancreas: diabetes mellitus I, up to 5% of patients;
- -
- Liver: hepatitis in up to 18% of cases that sometimes it can outcome in cirrhosis;
- -
- Stomach: chronic atrophic gastritis, with/without pernicious anemia, 13–27%. Patients having also intestinal metaplasia are at a high risk to develop gastric cancer;
- -
- Lung: pneumonitis, often misdiagnosed as bronchitis or asthma, due to the presence of cough;
- -
- Thyroid: Hashimoto thyroiditis;
- -
- Gut: malabsorption;
- -
- Kidneys: interstitial tubulonephritis and nephrolithiasis;
- -
- Spleen: asplenia;
- -
- Gonads: hypergonadotrophic hypogonadism, 24–60% of cases, as premature ovarian failure in females under 30 years of age;
- -
- Salivary and lacrimal glands (Sjogren’s like syndrome).
4.1. Differential Diagnosis
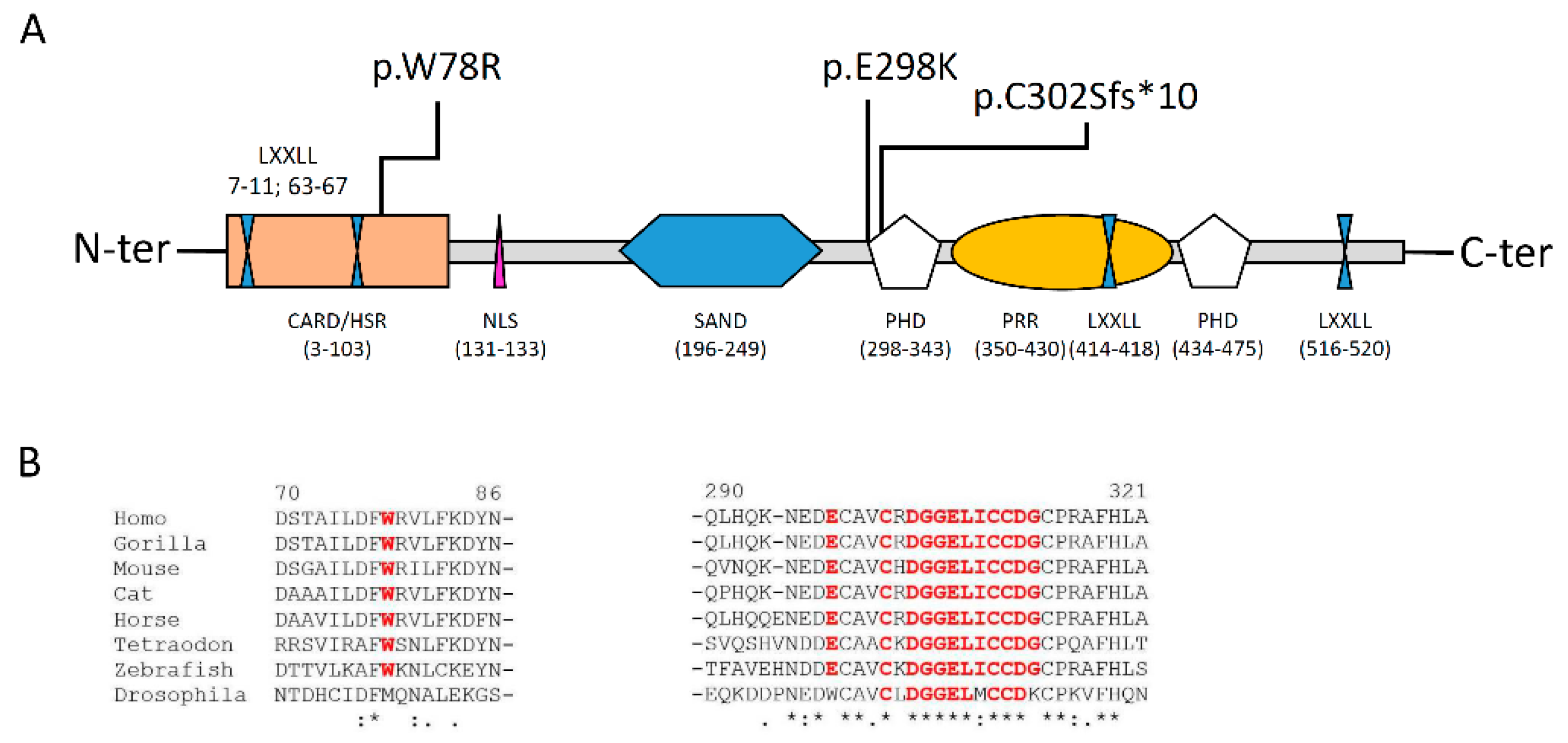
4.2. AIRE: Gene and Protein
- -
- One CARD/homogeneously staining region HSR (aa 3-103), involved in AIRE dimerization and in the chromatin binding;
- -
- Two zinc fingers of plant homeodomain (PHD1 and 2) type (aa 298-341 and 433-476, respectively) involved in the recruitment of chromatin related proteins and in AIRE transcriptional activity;
- -
- One DNA binding domain (SAND) that is engaged in promoting a protein–protein interaction with a transcriptional repressive complex;
- -
- Four nuclear receptor binding LXXLL modules, involved in promoting gene transcription;
- -
- One inverted LXXXLL domain, as transcription coactivator;
- -
- One proline rich region (PRR) involved in promoting gene transcription.
- -
- -
- It binds to different proteins, such positive elongation factors (P-TEFb) and heterogeneous ribonucleoproteins (HNRPL and RNA pol II) [31];
- -
- It is involved in the modulation of chromatin mediated by the interaction with a specific complex (ATF7IP-MBD1) in order to induce the CpG demethylation of TSAs genes [32].
4.3. Pathogenic Effect of the p.(Trp78Arg) and p.(Gly2898Lys) Variants
- i.
- Recessive, mostly located in the CARD/HSR domain with a direct impact on the homo-tetramerization of the complex; mutations falling in this group cause a “classic phenotype”;
- ii.
- Dominant-negative, mostly located in the PHD1 domain and affecting the transactivation-transcription function; mutations falling in this group cause a, often, milder “non-classical phenotype” [9].
4.4. Genetic Analysis as Helpful Tool for a Definitive Diagnosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
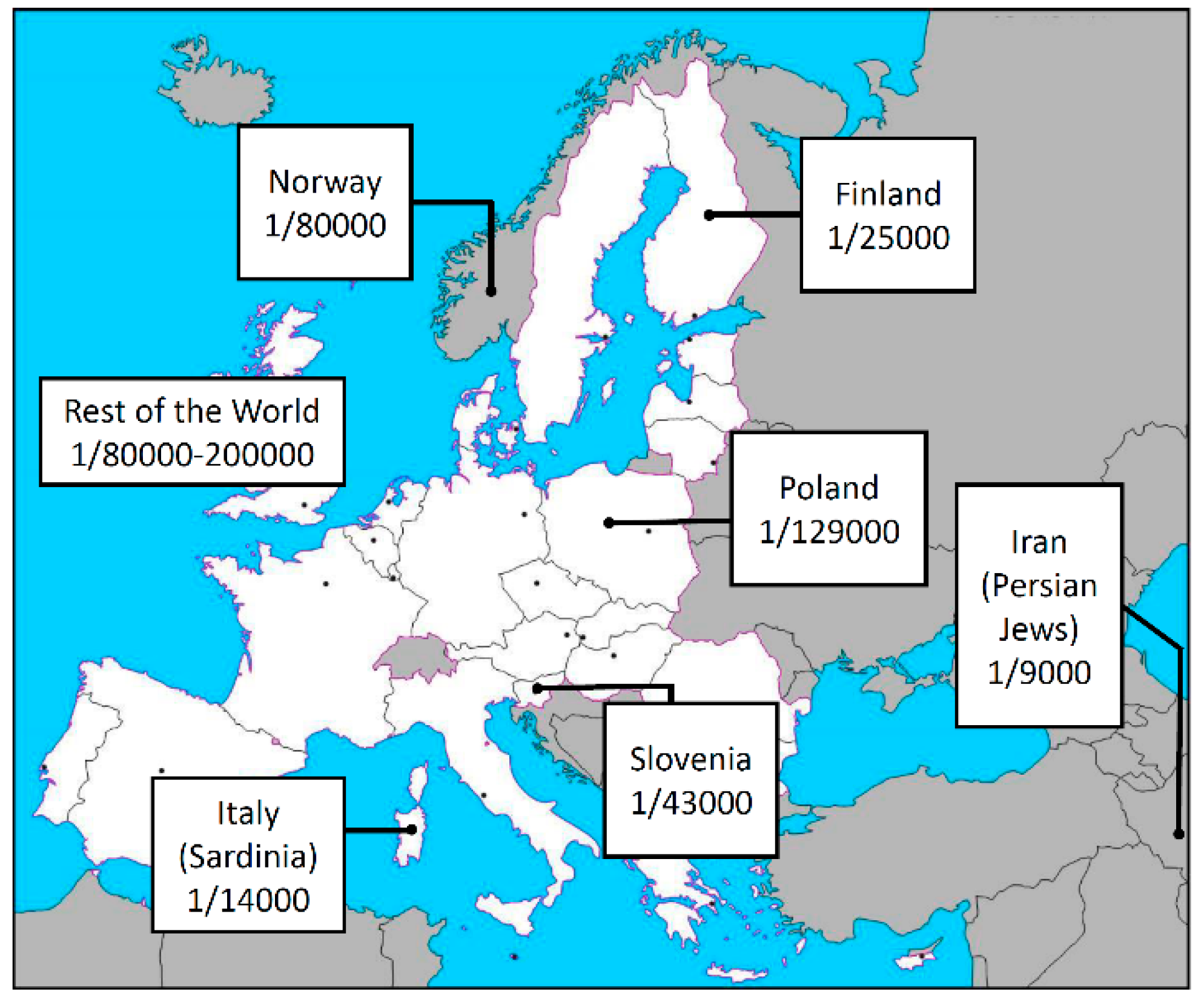
- Zlotogora, J.; Shapiro, M.S. Polyglandular autoimmune syndrome type I among Iranian Jews. J. Med. Genet. 1992, 29, 824–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosatelli, M.C.; Meloni, A.; Meloni, A.; Devoto, M.; Cao, A.; Scott, H.S.; Peterson, P.; Heino, M.; Krohn, K.J.; Nagamine, K.; et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum. Genet. 1998, 103, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.S.; Erichsen, M.M.; Meager, A.; Magitta, N.F.; Myhre, A.G.; Bollerslev, J.; Fougner, K.J.; Lima, K.; Knappskog, P.M.; Husebye, E.S. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J. Clin. Endocrinol. Metab. 2007, 92, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, Ø.; Oftedal, B.E.; Landegren, N.; Erichsen, M.M.; Bratland, E.; Lima, K.; Jørgensen, A.P.; Myhre, A.G.; Svartberg, J.; Fougner, K.J.; et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J. Clin. Endocrinol. Metab. 2016, 101, 2975–2983. [Google Scholar] [PubMed]
- Ahonen, P.; Myllärniemi, S.; Sipilä, I.; Perheentupa, J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APS-1) in a series of 68 patients. N. Engl. J. Med. 1990, 28, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Perheentupa, J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 2006, 91, 2843–2850. [Google Scholar] [CrossRef] [Green Version]
- Betterle, C.; Greggio, N.A.; Volpato, M. Clinical review 93: Autoimmune polyglandular syndrome type 1. J. Clin. Endocrinol. Metab. 1998, 83, 1049–1055. [Google Scholar] [CrossRef]
- Meager, A.; Visvalingam, K.; Peterson, P.; Möll, K.; Murumägi, A.; Krohn, K.; Eskelin, P.; Perheentupa, J.; Husebye, E.; Kadota, Y.; et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 2006, 3, e289. [Google Scholar] [CrossRef] [Green Version]
- Husebye, E.S.; Perheentupa, J.; Rautemaa, R.; Kämpe, O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med. 2009, 265, 514–529. [Google Scholar] [CrossRef]
- Nagamine, K.; Peterson, P.; Scott, H.S.; Kudoh, J.; Minoshima, S.; Heino, M.; Krohn, K.J.E.; Lalioti, M.D.; Mullis, P.E.; Antonarakis, S.E.; et al. Positional cloning of the APS-1 gene. Nat. Genet. 1997, 17, 393–398. [Google Scholar] [CrossRef]
- Chan, A.Y.; Anderson, M.S. Central tolerance to self revealed by the autoimmune regulator. Ann. N. Y. Acad. Sci. 2015, 1356, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, P.; Org, T.; Rebane, A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol. 2008, 8, 948–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oftedal, B.E.; Hellesen, A.; Erichsen, M.M.; Bratland, E.; Vardi, A.; Perheentupa, J.; Kemp, E.H.; Fiskerstrand, T.; Viken, M.K.; Weetman, A.P.; et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity 2015, 42, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.G.; Laloraya, M.; Wang, C.Y.; Ruan, Q.G.; Davoodi-Semiromi, A.; Kao, K.J.; She, J.X. The autoimmune regulator (AIRE) is a DNA-binding protein. J. Biol. Chem. 2001, 276, 41357–41364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martino, L.; Capalbo, D.; Improda, N.; Lorello, P.; Ungaro, C.; Di Mase, R.; Cirillo, E.; Pignata, C.; Salerno, M. Novel Findings into AIRE Genetics and Functioning: Clinical Implications. Front. Pediatr. 2016, 22, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rautemaa, R.; Hietanen, J.; Niissalo, S.; Pirinen, S.; Perheentupa, J. Oral and oesophageal squamous cell carcinoma--a complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I). Oral. Oncol. 2007, 6, 607–613. [Google Scholar] [CrossRef]
- Bensing, S.; Brandt, L.; Tabaroj, F.; Sjöberg, O.; Nilsson, B.; Ekbom, A.; Blomqvist, P.; Kämpe, O. Increased death risk and altered cancer incidence pattern in patients with isolated or combined autoimmune primary adrenocortical insufficiency. Clin. Endocrinol. 2008, 5, 697–704. [Google Scholar] [CrossRef]
- Alimohammadi, M.; Björklund, P.; Hallgren, A.; Pöntynen, N.; Szinnai, G.; Shikama, N.; Keller, M.P.; Ekwall, O.; Kinkel, S.A.; Husebye, E.S.; et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N. Engl. J. Med. 2008, 10, 1018–1028. [Google Scholar]
- Burk, C.J.; Ciocca, G.; Heath, C.R.; Duarte, A.; Dohil, M.; Connelly, E.A. Addison’s disease, diffuse skin, and mucosal hyperpigmenation with subtle “flu-like” symptoms—A report of two cases. Pediatr. Dermatol. 2008, 2, 215–218. [Google Scholar] [CrossRef]
- Winqvist, O.; Karlsson, F.A.; Kämpe, O. 21-Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 1992, 8809, 1559–1562. [Google Scholar] [CrossRef]
- Constantine, G.M.; Lionakis, M.S. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 2019, 287, 103–120. [Google Scholar] [CrossRef]
- Leibowitz, G.; Amir, G.; Losses, I.S.; Eliakim, R. Autoimmune polyglandular failure associated with malabsorption and gastric carcinoid tumour. J. Intern. Med. 1993, 6, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Ferre, E.M.N.; Rose, S.R.; Rosenzweig, S.D.; Burbelo, P.D.; Romito, K.R.; Niemela, J.E.; Rosen, L.B.; Break, T.J.; Gu, W.; Hunsberger, S.; et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insightig. 2016, 18, e88782. [Google Scholar] [CrossRef] [PubMed]
- Triolo, T.M.; Baschal, E.E.; Armstrong, T.K.; Toews, C.S.; Fain, P.R.; Rewers, M.J.; Yu, L.; Miao, D.; Eisenbarth, G.S.; Gottlieb, P.A.; et al. Homozygosity of the polymorphism MICA5.1 identifies extreme risk of progression to overt adrenal insufficiency among 21-hydroxylase antibody-positive patients with type 1 diabetes. J. Clin. Endocrinol. Metab. 2009, 11, 4517–4523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 12, 1132–1141. [Google Scholar]
- Gardner, J.M.; Metzger, T.C.; McMahon, E.J.; Au-Yeung, B.B.; Krawisz, A.K.; Lu, W.; Price, J.D.; Johannes, K.P.; Satpathy, A.T.; Murphy, K.M.; et al. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4+ T cells. Immunity 2013, 39, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, Y.; Hirota, F.; Yano, M.; Kitajima, H.; Miyazaki, J.; Kawamoto, H.; Mouri, Y.; Matsumoto, M. Biphasic Aire expression in early embryos and in medullary thymic epithelial cells before end-stage terminal differentiation. J. Exp. Med. 2010, 207, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar]
- Gotter, J.; Brors, B.; Hergenhahn, M.; Kyewski, B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J. Exp. Med. 2004, 199, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.S.; Su, M.A. AIRE expands: New roles in immune tolerance and beyond. Nat. Rev. Immunol. 2016, 16, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Waterfield, M.; Khan, I.S.; Cortez, J.T.; Fan, U.; Metzger, T.; Greer, A.; Fasano, K.; Martinez-Llordella, M.; Pollack, J.L.; Erle, D.J.; et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat. Immunol. 2014, 15, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Meloni, A.; Perniola, R.; Faà, V.; Corvaglia, E.; Cao, A.; Rosatelli, M.C. Delineation of the molecular defects in the AIRE gene in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients from Southern Italy. J. Clin. Endocrinol. Metab. 2002, 87, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Cervato, S.; Mariniello, B.; Lazzarotto, F.; Morlin, L.; Zanchetta, R.; Radetti, G.; De Luca, F.; Valenzise, M.; Giordano, R.; Rizzo, D.; et al. Evaluation of the autoimmune regulator (AIRE) gene mutations in a cohort of Italian patients with autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy (APS-1) and in their relatives. Clin. Endocrinol. 2009, 70, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Oftedal, B.E.; Wolff, A.B.; Husebye, E.S. AIRE-mutations and autoimmune disease. Curr. Opin. Immunol. 2016, 43, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Proekt, I.; Miller, C.N.; Lionakis, M.S.; Anderson, M.S. Insights into immune tolerance from AIRE deficiency. Curr. Opin. Immunol. 2017, 49, 71–78. [Google Scholar] [CrossRef]
- Betterle, C.; Ghizzoni, L.; Cassio, A.; Baronio, F.; Cervato, S.; Garelli, S.; Barbi, E.; Tonini, G. Autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy in Calabria: clinical, immunological and genetic patterns. J. Endocrinol. Investig. 2012, 35, 877–881. [Google Scholar]
- Capalbo, D.; Mazza, C.; Giordano, R.; Improda, N.; Arvat, E.; Cervato, S.; Morlin, L.; Pignata, C.; Betterle, C.; Salerno, M. Molecular background and genotype-phenotype correlation in autoimmune-polyendocrinopathy-candidiasis-ectodermal-distrophy patients from Campania and in their relatives. J. Endocrinol. Investig. 2012, 35, 169–173. [Google Scholar]
- Meloni, A.; Fiorillo, E.; Corda, D.; Perniola, R.; Cao, A.; Rosatelli, M.C. Two novel mutations of the AIRE protein affecting its homodimerization properties. Hum. Mutat. 2005, 25, 319. [Google Scholar] [CrossRef] [Green Version]
- Chignola, F.; Gaetani, M.; Rebane, A.; Org, T.; Mollica, L.; Zucchelli, C.; Spitaleri, A.; Mannella, V.; Peterson, P.; Musco, G. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009, 37, 2951–2961. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cinque, L.; Angeletti, C.; Orrico, A.; Castellana, S.; Ferrito, L.; Ciuoli, C.; Mazza, T.; Castori, M.; Guarnieri, V. Novel Pathogenic Variants of the AIRE Gene in Two Autoimmune Polyendocrine Syndrome Type I Cases with Atypical Presentation: Role of the NGS in Diagnostic Pathway and Review of the Literature. Biomedicines 2020, 8, 631. https://doi.org/10.3390/biomedicines8120631
Cinque L, Angeletti C, Orrico A, Castellana S, Ferrito L, Ciuoli C, Mazza T, Castori M, Guarnieri V. Novel Pathogenic Variants of the AIRE Gene in Two Autoimmune Polyendocrine Syndrome Type I Cases with Atypical Presentation: Role of the NGS in Diagnostic Pathway and Review of the Literature. Biomedicines. 2020; 8(12):631. https://doi.org/10.3390/biomedicines8120631
Chicago/Turabian StyleCinque, Luigia, Cristina Angeletti, Alfredo Orrico, Stefano Castellana, Lucia Ferrito, Cristina Ciuoli, Tommaso Mazza, Marco Castori, and Vito Guarnieri. 2020. "Novel Pathogenic Variants of the AIRE Gene in Two Autoimmune Polyendocrine Syndrome Type I Cases with Atypical Presentation: Role of the NGS in Diagnostic Pathway and Review of the Literature" Biomedicines 8, no. 12: 631. https://doi.org/10.3390/biomedicines8120631
APA StyleCinque, L., Angeletti, C., Orrico, A., Castellana, S., Ferrito, L., Ciuoli, C., Mazza, T., Castori, M., & Guarnieri, V. (2020). Novel Pathogenic Variants of the AIRE Gene in Two Autoimmune Polyendocrine Syndrome Type I Cases with Atypical Presentation: Role of the NGS in Diagnostic Pathway and Review of the Literature. Biomedicines, 8(12), 631. https://doi.org/10.3390/biomedicines8120631