STAT3 and p53: Dual Target for Cancer Therapy



Abstract
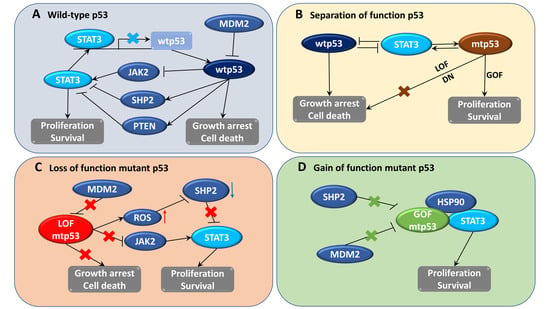
:
1. Introduction
2. Role of STAT3 Signaling in Cancer
2.1. Activation and Regulation of STAT3
2.2. Function as an Oncogene
2.3. Targeting STAT3 for Cancer Therapies
3. The Contribution of p53 in Cancer
3.1. Role of wtp53
3.2. Negative Regulation of wtp53
3.3. p53 Mutations in Cancer—From Loss of Function to Gain of Function
3.4. Mutant p53 and Cancer Therapy Resistance
4. STAT3 and p53 Feedback Regulation
4.1. Interaction between STAT3 and p53
4.2. STAT3 Inhibits p53-Mediated Apoptosis and Growth Arrest
4.3. p53 Regulates STAT3 Signaling in Cancer Cells
4.4. Constitutive Activation of STAT3 Occurs in Cancer Cells Containing p53 Mutations
5. Pharmacological Strategies to Target Both p53 and STAT3 Activities in Cancer Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APAF1 | Apoptotic peptidase activating factor 1 |
| BAX | BCL2 associated X |
| DDB2 | Damage specific DNA binding protein 2 |
| E2F7 | E2F transcription factor 7 |
| ERCC5 | ERCC excision repair 5 |
| FANCC | FA complementation group C |
| GADD45 | Growth arrest and DNA damage inducible |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| MSH2 | MutS homolog 2 |
| PML | Promyelocytic leukemia |
| POLK/POLH | DNA polymerase kappa/ DNA polymerase eta |
| PUMA | p53 upregulated modulator of apoptosis |
| SLC7A11 | Solute carrier family 7 member 11 |
| TIGAR | TP53 induced glycolysis regulatory phosphatase |
| XPC | Xeroderma pigmentosum, complementation group C |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummar, S.; Gutierrez, M.; Doroshow, J.H.; Murgo, A.J. Drug development in oncology: Classical cytotoxics and molecularly targeted agents. Br. J. Clin. Pharmacol. 2006, 62, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Lieblein, J.C.; Ball, S.; Hutzen, B.; Sasser, A.K.; Lin, H.J.; Huang, T.H.; Hall, B.M.; Lin, J. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 2008, 8, 302. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Jin, Z.; Liu, Z.; Sun, L.; Wang, L.; Li, K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol. Diagn. Ther. 2011, 15, 347–352. [Google Scholar] [CrossRef]
- Xu, Y.H.; Lu, S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur. J. Surg. Oncol. 2014, 40, 311–317. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsieh, F.C.; Lieblein, J.C.; Brown, J.; Chan, C.; Wallace, J.A.; Cheng, G.; Hall, B.M.; Lin, J. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 2007, 96, 591–599. [Google Scholar] [CrossRef]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarese, T.M.; Campbell, C.L.; McQuain, C.; Mitchell, K.; Guardiani, R.; Quesenberry, P.J.; Nelson, B.E. Coexpression of oncostatin M and its receptors and evidence for STAT3 activation in human ovarian carcinomas. Cytokine 2002, 17, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Kanda, N.; Seno, H.; Konda, Y.; Marusawa, H.; Kanai, M.; Nakajima, T.; Kawashima, T.; Nanakin, A.; Sawabu, T.; Uenoyama, Y.; et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene 2004, 23, 4921–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeong, A.J.; Ye, S.K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Song, D.; Li, H.; Yang, Y.; Ma, X.; Deng, S.; Ren, C.; Shu, X. Negative regulators of STAT3 signaling pathway in cancers. Cancer Manag. Res. 2019, 11, 4957–4969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Royds, J.A.; Iacopetta, B. p53 and disease: When the guardian angel fails. Cell Death Differ. 2006, 13, 1017–1026. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Tang, H.; Jin, X.; Jia, G.; Hsieh, J.T. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene 2002, 21, 3082–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Jin, X.; Rothman, K.; Lin, H.J.; Tang, H.; Burke, W. Modulation of signal transducer and activator of transcription 3 activities by p53 tumor suppressor in breast cancer cells. Cancer Res. 2002, 62, 376–380. [Google Scholar] [PubMed]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene—Still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Tweardy, D.J.; Zhang, M.; Zhang, X.; Landua, J.; Petrovic, I.; Bu, W.; Roarty, K.; Hilsenbeck, S.G.; Rosen, J.M.; et al. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 2014, 32, 2571–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef] [Green Version]
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Gorgulu, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [Green Version]
- Spehlmann, M.E.; Manthey, C.F.; Dann, S.M.; Hanson, E.; Sandhu, S.S.; Liu, L.Y.; Abdelmalak, F.K.; Diamanti, M.A.; Retzlaff, K.; Scheller, J.; et al. Trp53 Deficiency Protects against Acute Intestinal Inflammation. J. Immunol. 2013, 191, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, H.; Lin, H.J.; Yang, S.; Lin, J.; Liang, G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol. Sci. 2016, 37, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.H.; Hafeez, B.B.; Sand, J.M.; Pierce, D.B.; Aziz, S.W.; Dreckschmidt, N.E.; Verma, A.K. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene 2010, 29, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Bak, Y.; Oh, J.W.; Hong, J.; Lee, S.; Hong, J.T.; Yoon, D.Y. Inhibition of IL-13 and IL-13Ralpha2 Expression by IL-32theta in Human Monocytic Cells Requires PKCdelta and STAT3 Association. Int. J. Mol. Sci. 2019, 20, 1949. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Sun, S.; Steinberg, B.M. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J. Gen. Virol. 2002, 83 Pt 7, 1651–1658. [Google Scholar] [CrossRef] [Green Version]
- Irie-Sasaki, J.; Sasaki, T.; Matsumoto, W.; Opavsky, A.; Cheng, M.; Welstead, G.; Griffiths, E.; Krawczyk, C.; Richardson, C.D.; Aitken, K.; et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 2001, 409, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Li, S.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, W.S.; Hilton, D.J. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004, 22, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by pias proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Rincon, M. Mitochondrial Stat3, the Need for Design Thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Oka, M.; Iwasaki, T.; Fukami, Y.; Nishigori, C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J. Investig. Dermatol. 2012, 132, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarajan, E.; Huang, S. STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 2009, 9, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Inghirami, G. STAT3: A multifaceted oncogene. Proc. Natl. Acad. Sci. USA 2006, 103, 10151–10152. [Google Scholar] [CrossRef] [Green Version]
- Spiotto, M.T.; Chung, T.D. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate 2000, 42, 186–195. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, U.; Kasembeli, M.M.; Tweardy, D.J. STAT3 inhibitors in cancer: A comprehensive update. In STAT Inhibitors in Cancer; Springer: New York, NY, USA, 2016; pp. 95–161. [Google Scholar]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Wang, G.; Li, W.; Hu, X.; Huang, Q.; Xu, K.; Lou, W.; Wu, J.; Liang, C.; Lou, Q.; et al. Activation of the JAK-STAT3 pathway is associated with the growth of colorectal carcinoma cells. Oncol. Rep. 2014, 31, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Jove, V.; Buettner, R.; Xin, H.; Wu, J.; Wang, Y.; Nam, S.; Xu, Y.; Ara, T.; DeClerck, Y.A.; et al. Sorafenib inhibits endogenous and IL-6/S1P induced JAK2-STAT3 signaling in human neuroblastoma, associated with growth suppression and apoptosis. Cancer Biol. Ther. 2012, 13, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Konig, H.; Holyoake, T.L.; Bhatia, R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood 2008, 111, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Devgan, G.; Darnell, J.E., Jr.; Bromberg, J.F. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hedou, D.; Viaud-Massuard, M.C.; Prie, G.; Gouilleux, F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Shirole, N.; Tian, R.; Pal, D.; Sordella, R. The Evolution of TP53 Mutations: From Loss-of-Function to Separation-of-Function Mutants. J. Cancer Biol. Res. 2016, 4, 1091. [Google Scholar] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Shirole, N.H.; Pal, D.; Kastenhuber, E.R.; Senturk, S.; Boroda, J.; Pisterzi, P.; Miller, M.; Munoz, G.; Anderluh, M.; Ladanyi, M.; et al. TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife 2016, 5, e17929. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K. The Contrived Mutant p53 Oncogene—Beyond Loss of Functions. Front. Oncol. 2015, 5, 276. [Google Scholar] [CrossRef]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugieres, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Huang, J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol. Ther. 2020, 107720. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Fontemaggi, G.; Strano, S.; Blandino, G.; D’Orazi, G. Targeting mutant p53 in cancer: The latest insights. J. Exp. Clin. Cancer Res. 2019, 38, 290. [Google Scholar] [CrossRef] [PubMed]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalniak, L.; Kocik, J.; Polak, J.; Skalniak, A.; Rak, M.; Wolnicka-Glubisz, A.; Holak, T.A. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers 2018, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [Green Version]
- Berberich, A.; Kessler, T.; Thome, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.G.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.C.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef]
- Polotskaia, A.; Xiao, G.; Reynoso, K.; Martin, C.; Qiu, W.G.; Hendrickson, R.C.; Bargonetti, J. Proteome-wide analysis of mutant p53 targets in breast cancer identifies new levels of gain-of-function that influence PARP, PCNA, and MCM4. Proc. Natl. Acad. Sci. USA 2015, 112, E1220–E1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, M.K.; Movassagh, M.; Siggens, L.; Vujic, A.; Goddard, M.; Sanchez, A.; Perkins, N.; Figg, N.; Bennett, M.; Carroll, J.; et al. High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-kappaB-complex-dependent gene expression in human heart failure. Genome Med. 2010, 2, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, L.E.; Wang, Y.; Zhong, M.; Wang, D.; Chakka, A.B.; Yang, Z.; Li, F.; Song, Q.; Rigatti, L.H.; Chaparala, S.; et al. EAF2 and p53 Co-Regulate STAT3 Activation in Prostate Cancer. Neoplasia 2018, 20, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Eves, R.; Jia, L.; Mooney, P.; Mak, A.S. p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol. Cell. Biol. 2009, 29, 3088–3098. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, U.K.; Mooney, P.; Jia, L.; Eves, R.; Raptis, L.; Mak, A.S. Doubles game: Src-Stat3 versus p53-PTEN in cellular migration and invasion. Mol. Cell. Biol. 2010, 30, 4980–4995. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Piipponen, M.; Nissinen, L.; Riihila, P.; Farshchian, M.; Kallajoki, M.; Peltonen, J.; Peltonen, S.; Kahari, V.M. p53-Regulated Long Noncoding RNA PRECSIT Promotes Progression of Cutaneous Squamous Cell Carcinoma via STAT3 Signaling. Am. J. Pathol. 2020, 190, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, B.; Tian, Y.; Dan, J.; Luo, Y.; Wu, X. P53 Mutant p53(N236S) Regulates Cancer-Associated Fibroblasts Properties Through Stat3 Pathway. OncoTargets Ther. 2020, 13, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Edsbacker, E.; Serviss, J.T.; Kolosenko, I.; Palm-Apergi, C.; De Milito, A.; Tamm, K.P. STAT3 is activated in multicellular spheroids of colon carcinoma cells and mediates expression of IRF9 and interferon stimulated genes. Sci. Rep. 2019, 9, 536. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, N.R.; Rowan, A.; Smith, M.E.; Kerr, I.B.; Bodmer, W.F.; Gannon, J.V.; Lane, D.P. p53 mutations in colorectal cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 7555–7559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knosel, T.; Kenner, L.; et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bodmer, W.F. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Lai, R.; Chirieac, L.R.; Li, C.; Thomazy, V.A.; Grammatikakis, I.; Rassidakis, G.Z.; Zhang, W.; Fujio, Y.; Kunisada, K.; et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: Inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am. J. Pathol. 2005, 167, 969–980. [Google Scholar] [CrossRef]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Pan, J.; Ji, L.; Song, S.; Lv, H.; Yang, Z.; Guo, Y. Carboxypeptidase A4 promotes cell growth via activating STAT3 and ERK signaling pathways and predicts a poor prognosis in colorectal cancer. Int. J. Biol. Macromol. 2019, 138, 125–134. [Google Scholar] [CrossRef]
- Yun, H.J.; Kim, S.Y.; Kwon, Y.Y.; Kim, C.H.; Kang, C.M.; Kim, E.J. Janus-activated kinases and signal transducer and activator of transcription control tumor growth response to camptothecin in human colon cancer cells. Cancer Biol. Ther. 2010, 10, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.J.; Li, P.K.; Li, C.; Lin, J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57, 4285–4300. [Google Scholar] [PubMed]
- Lamy, V.; Bousserouel, S.; Gosse, F.; Minker, C.; Lobstein, A.; Raul, F. p53 Activates Either Survival or Apoptotic Signaling Responses in Lupulone-Treated Human Colon Adenocarcinoma Cells and Derived Metastatic Cells. Transl. Oncol. 2010, 3, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasielewski, M.; Elstrodt, F.; Klijn, J.G.; Berns, E.M.; Schutte, M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res. Treat. 2006, 99, 97–101. [Google Scholar] [CrossRef]
- Chen, M.; Pockaj, B.; Andreozzi, M.; Barrett, M.T.; Krishna, S.; Eaton, S.; Niu, R.; Anderson, K.S. JAK2 and PD-L1 Amplification Enhance the Dynamic Expression of PD-L1 in Triple-negative Breast Cancer. Clin. Breast Cancer 2018, 18, e1205–e1215. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Dhillon, J.; Wang, M.Y.; Gao, Y.; Hu, K.; Park, E.; Astanehe, A.; Hung, M.C.; Eirew, P.; Eaves, C.J.; et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008, 68, 8661–8666. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Saxena, R.; Asif, N.; Sinclair, E.; Tan, J.; Cruz, I.; Berry, D.; Kallakury, B.; Pham, Q.; Wang, T.T.Y.; et al. p53 mutant-type in human prostate cancer cells determines the sensitivity to phenethyl isothiocyanate induced growth inhibition. J. Exp. Clin. Cancer Res. 2019, 38, 307. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Cho, S.G.; Choi, Y.J.; Yun, Y.J.; Lee, K.M.; Lee, K.; Yoo, H.H.; Shin, Y.C.; Ko, S.G. SH003 suppresses breast cancer growth by accumulating p62 in autolysosomes. Oncotarget 2017, 8, 88386–88400. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol. Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C.D. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akca, H.; Tani, M.; Hishida, T.; Matsumoto, S.; Yokota, J. Activation of the AKT and STAT3 pathways and prolonged survival by a mutant EGFR in human lung cancer cells. Lung Cancer 2006, 54, 25–33. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Liu, Y.; Zhang, R.; Zhang, B.; Wang, T.; Zhu, X.; Mei, L.; Chen, H.; Zhang, H.; Ming, P.; et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal pathway in Gefitinib-resistant lung cancer cells. Sci. Rep. 2015, 5, 8477. [Google Scholar] [CrossRef]
- Carr, A.C.; Khaled, A.S.; Bassiouni, R.; Flores, O.; Nierenberg, D.; Bhatti, H.; Vishnubhotla, P.; Manuel, J.P.; Santra, S.; Khaled, A.R. Targeting chaperonin containing TCP1 (CCT) as a molecular therapeutic for small cell lung cancer. Oncotarget 2017, 8, 110273–110288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wu, J.; Shi, J.; Huo, Y.M.; Dai, W.; Geng, J.; Lu, P.; Yang, M.W.; Fang, Y.; Wang, W.; et al. IL22RA1/STAT3 Signaling Promotes Stemness and Tumorigenicity in Pancreatic Cancer. Cancer Res. 2018, 78, 3293–3305. [Google Scholar]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. ZEB1 and IL-6/11-STAT3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of S100 proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar] [PubMed]
- Mullany, L.K.; Wong, K.K.; Marciano, D.C.; Katsonis, P.; King-Crane, E.R.; Ren, Y.A.; Lichtarge, O.; Richards, J.S. Specific TP53 Mutants Overrepresented in Ovarian Cancer Impact CNV, TP53 Activity, Responses to Nutlin-3a, and Cell Survival. Neoplasia 2015, 17, 789–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saydmohammed, M.; Joseph, D.; Syed, V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in ovarian and endometrial cancer cells. J. Cell. Biochem. 2010, 110, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, Y.; Westphal, H. Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res. 1992, 52, 4196–4199. [Google Scholar] [PubMed]
- Huang, M.; Page, C.; Reynolds, R.K.; Lin, J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecol. Oncol. 2000, 79, 67–73. [Google Scholar] [CrossRef]
- Skilling, J.S.; Squatrito, R.C.; Connor, J.P.; Niemann, T.; Buller, R.E. p53 gene mutation analysis and antisense-mediated growth inhibition of human ovarian carcinoma cell lines. Gynecol. Oncol. 1996, 60, 72–80. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Srivastava, S.K. Critical role of STAT3 in melanoma metastasis through anoikis resistance. Oncotarget 2014, 5, 7051–7064. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Sun, Y.; Guo, Y.; Qin, G.; Mu, S.; Fan, R.; Wang, B.; Gao, W.; Wu, H.; Wang, G.; et al. NF-YA promotes invasion and angiogenesis by upregulating EZH2-STAT3 signaling in human melanoma cells. Oncol. Rep. 2016, 35, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 signaling in acute myeloid leukemia: Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef]
- Pathania, A.S.; Kumar, S.; Guru, S.K.; Bhushan, S.; Sharma, P.R.; Aithagani, S.K.; Singh, P.P.; Vishwakarma, R.A.; Kumar, A.; Malik, F. The Synthetic Tryptanthrin Analogue Suppresses STAT3 Signaling and Induces Caspase Dependent Apoptosis via ERK Up Regulation in Human Leukemia HL-60 Cells. PLoS ONE 2014, 9, e110411. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: Implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Kim, C.; Lee, J.; Um, J.Y.; Sethi, G.; Ahn, K.S. Arctiin is a pharmacological inhibitor of STAT3 phosphorylation at tyrosine 705 residue and potentiates bortezomib-induced apoptotic and anti-angiogenic effects in human multiple myeloma cells. Phytomedicine 2019, 55, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Yuzugullu, H.; Von, T.; Thorpe, L.M.; Walker, S.R.; Roberts, T.M.; Frank, D.A.; Zhao, J.J. NTRK2 activation cooperates with PTEN deficiency in T-ALL through activation of both the PI3K-AKT and JAK-STAT3 pathways. Cell Discov. 2016, 2, 16030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Coronel, L.; Somalanka, B.; Raju, A.; Aning, O.A.; An, O.; Ho, Y.S.; Chen, S.; Mak, S.Y.; Hor, P.Y.; et al. Mitochondrial uncoupling reveals a novel therapeutic opportunity for p53-defective cancers. Nat. Commun. 2018, 9, 3931. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 1102. [Google Scholar] [CrossRef]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, R.; Carillo, V.; Romeo, M.A.; Gaeta, A.; Nazzari, C.; Gonnella, R.; Granato, M.; D’Orazi, G.; Faggioni, A.; Cirone, M. STAT3 phosphorylation affects p53/p21 axis and KSHV lytic cycle activation. Virology 2019, 528, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, M.A.; El-Sherbeeny, N.A.; Helmi, N.; Alazragi, R.; Salem, N.; Elaidy, S.M. Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: Implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol. Cell. Biochem. 2020, 469, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, C.; Liu, Z.; Peng, F.; Chen, X.; Yang, G.; Zhang, D.; Yin, Z.; Ma, J.; Zheng, Z.; et al. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yue, X.; Zhao, Y.; Li, X.; Wu, L.; Zhang, C.; Liu, Z.; Lin, K.; Xu-Monette, Z.Y.; Young, K.H.; et al. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat. Commun. 2014, 5, 5218. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, J.K.; Yoo, J.Y. NFkappaB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Mol. Oncol. 2014, 8, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.B.; Hu, G.Y.; Long, G.X.; Qiu, H.; Mei, Q.; Hu, G.Q. Celecoxib induces apoptosis and cell-cycle arrest in nasopharyngeal carcinoma cell lines via inhibition of STAT3 phosphorylation. Acta Pharmacol. Sin. 2012, 33, 682–690. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, A.; Zhao, Z.; Xu, Y.; Lin, J.; Jou, D.; Li, C. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): Identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J. Med. Chem. 2011, 54, 5592–5596. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Lee, J.I.; Jin, D.H.; Lee, W.J.; Park, G.B.; Kim, S.; Kim, Y.S.; Wu, T.C.; Hur, D.Y.; Kim, D. Sequential treatment of HPV E6 and E7-expressing TC-1 cells with bortezomib and celecoxib promotes apoptosis through p-p38 MAPK-mediated downregulation of cyclin D1 and CDK2. Oncol. Rep. 2014, 31, 2429–2437. [Google Scholar] [CrossRef]
- Swamy, M.V.; Herzog, C.R.; Rao, C.V. Inhibition of COX-2 in colon cancer cell lines by celecoxib increases the nuclear localization of active p53. Cancer Res. 2003, 63, 5239–5242. [Google Scholar] [PubMed]
- Xu, X.T.; Hu, W.T.; Zhou, J.Y.; Tu, Y. Celecoxib enhances the radiosensitivity of HCT116 cells in a COX-2 independent manner by up-regulating BCCIP. Am. J. Transl. Res. 2017, 9, 1088–1100. [Google Scholar] [PubMed]
- Moon, H.J.; Kim, H.B.; Lee, S.H.; Jeun, S.E.; Kang, C.D.; Kim, S.H. Sensitization of multidrug-resistant cancer cells to Hsp90 inhibitors by NSAIDs-induced apoptotic and autophagic cell death. Oncotarget 2018, 9, 11303–11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ung, N.; Putoczki, T.L.; Stylli, S.S.; Ng, I.; Mariadason, J.M.; Chan, T.A.; Zhu, H.J.; Luwor, R.B. Anti-EGFR therapeutic efficacy correlates directly with inhibition of STAT3 activity. Cancer Biol. Ther. 2014, 15, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Benavente, S.; Armstrong, E.A.; Li, C.; Wheeler, D.L.; Harari, P.M. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2011, 71, 7071–7079. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Jin, M.; Ye, S.; Yan, S. CHI3L1 promotes proliferation and improves sensitivity to cetuximab in colon cancer cells by down-regulating p53. J. Clin. Lab. Anal. 2020, 34, e23026. [Google Scholar] [CrossRef]
- Narvi, E.; Vaparanta, K.; Karrila, A.; Chakroborty, D.; Knuutila, S.; Pulliainen, A.; Sundvall, M.; Elenius, K. Different responses of colorectal cancer cells to alternative sequences of cetuximab and oxaliplatin. Sci. Rep. 2018, 8, 16579. [Google Scholar] [CrossRef]
- Dudgeon, C.; Peng, R.; Wang, P.; Sebastiani, A.; Yu, J.; Zhang, L. Inhibiting oncogenic signaling by sorafenib activates PUMA via GSK3beta and NF-kappaB to suppress tumor cell growth. Oncogene 2012, 31, 4848–4858. [Google Scholar] [CrossRef] [Green Version]
- Roy Choudhury, S.; Karmakar, S.; Banik, N.L.; Ray, S.K. Synergistic efficacy of sorafenib and genistein in growth inhibition by down regulating angiogenic and survival factors and increasing apoptosis through upregulation of p53 and p21 in malignant neuroblastoma cells having N-Myc amplification or non-amplification. Investig. New Drugs 2010, 28, 812–824. [Google Scholar]
- Panka, D.J.; Liu, Q.; Geissler, A.K.; Mier, J.W. Effects of HDM2 antagonism on sunitinib resistance, p53 activation, SDF-1 induction, and tumor infiltration by CD11b+/Gr-1+ myeloid derived suppressor cells. Mol. Cancer 2013, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Xu, L.; Zhang, J.; Hu, X.; Liu, Y.; Yin, H.; Lv, T.; Zhang, H.; Liu, L.; An, H.; et al. Sunitinib induces cellular senescence via p53/Dec1 activation in renal cell carcinoma cells. Cancer Sci. 2013, 104, 1052–1061. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Heath, E.I.; Posadas, E.M.; Yu, E.Y.; Harrison, M.R.; Bruce, J.Y.; Cho, S.Y.; Wilding, G.E.; Fetterly, G.J.; Hangauer, D.G.; et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 71, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.S.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, U.; Eckols, T.K.; Kolosov, M.; Kasembeli, M.M.; Adam, A.; Torres, D.; Zhang, X.; Dobrolecki, L.E.; Wei, W.; Lewis, M.T.; et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene 2015, 34, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, W.; Yin, Z.X.; Liu, G.; Zeng, Q.; Shen, X.F.; Sun, Q.H.; Li, D.D.; Jian, Y.P.; Zhang, Y.H.; Wang, Y.S.; et al. Piperlongumine and p53-reactivator APR-246 selectively induce cell death in HNSCC by targeting GSTP1. Oncogene 2018, 37, 3384–3398. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumin, N.H.; Drobnitzky, N.; Patel, A.; Lourenco, L.M.; Cahill, F.F.; Jiang, Y.; Kong, A.; Ryan, A.J. Overcoming acquired resistance to HSP90 inhibition by targeting JAK-STAT signalling in triple-negative breast cancer. BMC Cancer 2019, 19, 102. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Mezina, A.; Shaib, W.L.; Landry, J.; El-Rayes, B.F. Targeting the Janus-activated kinase-2-STAT3 signalling pathway in pancreatic cancer using the HSP90 inhibitor ganetespib. Eur. J. Cancer 2016, 52, 109–119. [Google Scholar] [CrossRef]
- Spiegelberg, D.; Dascalu, A.; Mortensen, A.C.; Abramenkovs, A.; Kuku, G.; Nestor, M.; Stenerlow, B. The novel HSP90 inhibitor AT13387 potentiates radiation effects in squamous cell carcinoma and adenocarcinoma cells. Oncotarget 2015, 6, 35652–35666. [Google Scholar] [CrossRef]
- Chan, K.C.; Ting, C.M.; Chan, P.S.; Lo, M.C.; Lo, K.W.; Curry, J.E.; Smyth, T.; Lee, A.W.; Ng, W.T.; Tsao, G.S.; et al. A novel Hsp90 inhibitor AT13387 induces senescence in EBV-positive nasopharyngeal carcinoma cells and suppresses tumor formation. Mol. Cancer 2013, 12, 128. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Sung, K.S.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. HSP90 inhibitor NVP-AUY922 enhances TRAIL-induced apoptosis by suppressing the JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells. Cell Signal. 2015, 27, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuhmer, T.; Zollinger, A.; Siegmund, D.; Chatterjee, M.; Grella, E.; Knop, S.; Kortum, M.; Unzicker, C.; Jensen, M.R.; Quadt, C.; et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008, 22, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Chakraborty, S.N.; Leng, X.; Ma, H.; Arlinghaus, R.B. HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex in leukemia cells. Genes Cancer 2015, 6, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Best, O.G.; Mulligan, S.P. Heat shock protein-90 inhibitor, NVP-AUY922, is effective in combination with fludarabine against chronic lymphocytic leukemia cells cultured on CD40L-stromal layer and inhibits their activated/proliferative phenotype. Leuk. Lymphoma 2012, 53, 2314–2320. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tong, X.; Sun, L.; Li, H.; Jones, R.D.; Liao, J.; Yang, G.Y. Inhibition of mutant Kras and p53-driven pancreatic carcinogenesis by atorvastatin: Mainly via targeting of the farnesylated DNAJA1 in chaperoning mutant p53. Mol. Carcinog. 2019, 58, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Sharma, S.; Kumar, B.; Teknos, T.N. Atorvastatin inhibits RhoC function and limits head and neck cancer metastasis. Oral Oncol. 2013, 49, 778–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Huang, H.; Farischon, C.; Li, D.; Du, Z.; Zhang, K.; Zheng, X.; Goodin, S. Combined effects of atorvastatin and aspirin on growth and apoptosis in human prostate cancer cells. Oncol. Rep. 2017, 37, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.T.; Huang, S.W.; Liu, K.T.; Lee, T.Y.; Shieh, J.J.; Wu, C.Y. Atorvastatin-induced senescence of hepatocellular carcinoma is mediated by downregulation of hTERT through the suppression of the IL-6/STAT3 pathway. Cell Death Discov. 2020, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Tang, Y.; Fang, J.; Zhou, Z.; Xing, Z.; Guo, Z.; Guo, X.; Wang, W.; Jiao, W.; Xu, Z.; et al. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PLoS ONE 2013, 8, e62823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]


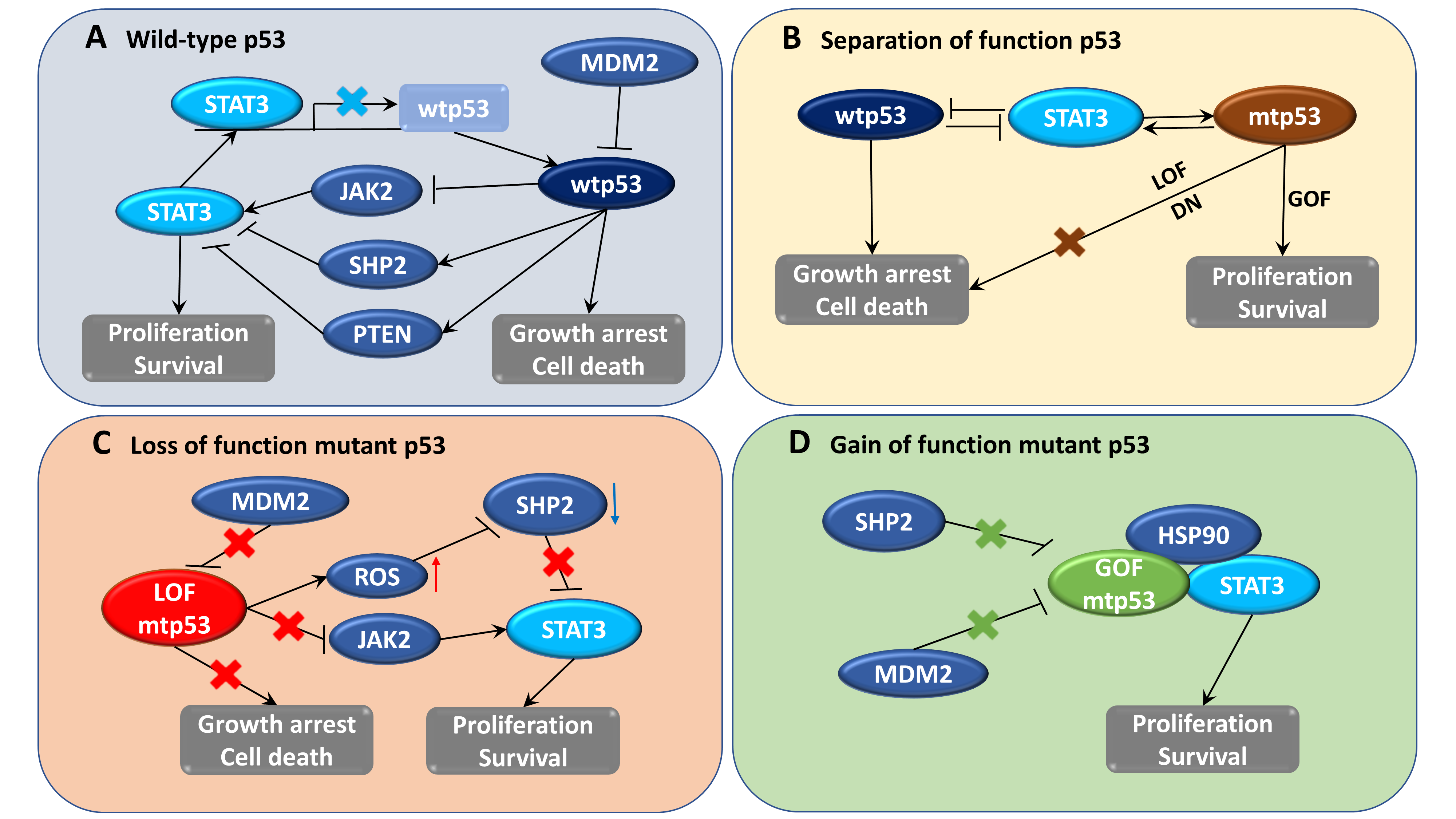{kind=link}
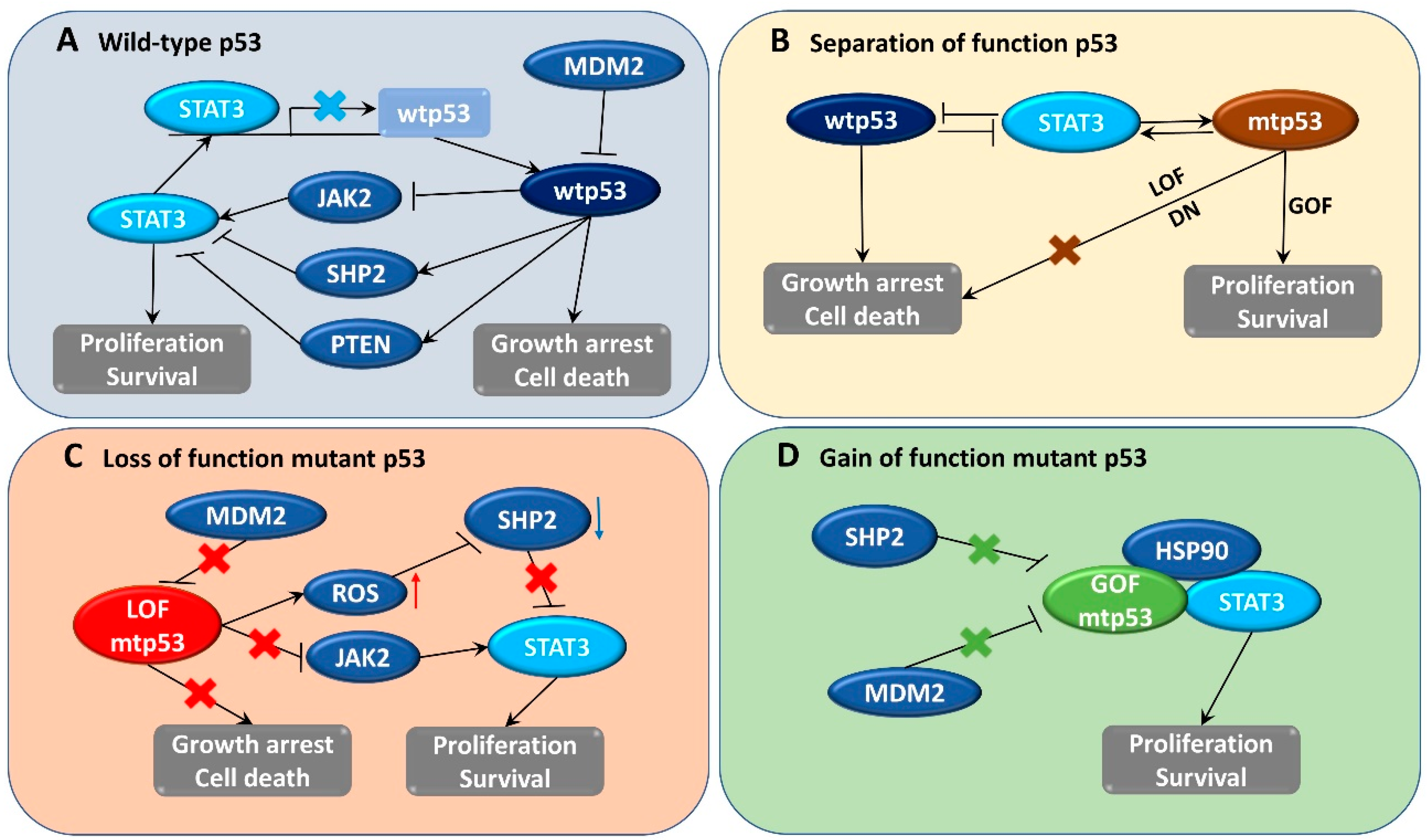{kind=link}
| Cancer Types | Cell Lines | p53 Status | STAT3 Status | Refs. |
|---|---|---|---|---|
| Colon | LS174T | Wild-type | No | [112,113] |
| SW48 | Wild-type | No | [114,115] | |
| SW480 | Mutant (R273H, P309S) a | Active | [28,116,117] | |
| LS123 | Mutant (R175H) a | Active | [118] | |
| LS1034 | Mutant (G245S) | Active | [28] | |
| COLO320DM | Mutant (R248W) | Active | [28] | |
| WiDr | Mutant (R273H) | Low | [112,119] | |
| HCT116 | Wild-type | Active | [115,120,121] | |
| HT-29 | Mutant (R273H) | Active | [116,117,122] | |
| SW620 | Mutant (R273H, P309S) a | No | [121,123] | |
| SW1463 | Mutant (R248Q) a | Active | [28] | |
| SW837 | Mutant (R248W) a | Active | [28] | |
| DLD-1 | Mutant (S241F) | No | [28,121] | |
| Breast | MDA-MB-231 | Mutant (R280K) a | Active | [38,39,124] |
| MDA-MB-361 | Mutant (E56X) | No | [38,125] | |
| MCF-7 | Wild type | Active | [38,39,126] | |
| MDA-MB-453 | Mutant (T387S) | No | [38,126,127] | |
| MDA-MB-435S | Mutant (G266E) | Active | [38,125] | |
| MDA-MB-468 | Mutant (R273H) a | Active | [124,125,127] | |
| MDA-MB-436 | Mutant (E204fsX45) | Active | [125,126] | |
| AU565 | Mutant (R175H) a | Active | [121,128] | |
| SK-BR-3 | Mutant (R175H) a | Active | [38,125,129] | |
| HCC70 | Mutant (R248Q) | Active | [126,130,131] | |
| BT-549 | Mutant (R249S) a | Active | [125,126] | |
| HCC38 | Mutant (R273L) | Active | [130,132,133] | |
| T47D | Mutant (L194F) a | Active | [38,125,126] | |
| Lung | BEAS-2B | Wild type b | No | [134] |
| A549 | Wild type | Active | [121,135] | |
| NCI-H596 | Mutant (G245C) | Active | [136] | |
| NCI-H1299 | Null | No | [135] | |
| NCI-H1975 | Mutant (R273H) | Active | [130,137] | |
| NCI-H1882 | Wild type b | Active | [138] | |
| NCI-H1417 | Mutant (R175L) b | Active | [138] | |
| NCI-H719 | Mutant (R248Q) b | Active | [138] | |
| NCI-H1105 | Mutant (R249S) b | Active | [138] | |
| NCI-H1048 | Mutant (R273C) b | Active | [130,138] | |
| Pancreatic | SW1990 | Wild type b | Active | [139] |
| SU.86.86 | Mutant (G245S) b | No | [140] | |
| BXPC-3 | Mutant (Y220C) a | No | [140] | |
| PANC-1 | Mutant (R273H) a | Active | [39,124] | |
| MIA-PaCa-2 | Mutant (R248W/R273H) a | Active | [140] | |
| Colo 357 | Wild-type | Active | [124] | |
| Prostate | DU-145 | Mutant (P223L/V274F) a | Active | [129,141] |
| PC-3 | Null | Active | [129,141] | |
| LNCaP | Wild-type | Active | [129,141] | |
| Ovary | SKOV3 | Mutant (H179R) | Active | [122,136,142,143] |
| OVCAR3 | Mutant (R248Q) | Active | [142,143] | |
| OVCA420 | Mutant (R273H) | Active | [142,144] | |
| OVCA433 | Mutant (E258K) | Active | [142,145] | |
| OVCA429 | Wild-type | Active | [144] | |
| Caov-3 | Mutant (Q136 c) | Active | [146,147] | |
| A2780 | Wild-type | No | [147,148] | |
| MDAH 2774 | Mutant (R273H) | Active | [147,148] | |
| SW626 | Mutant (G262V) b | No | [147] | |
| Melanoma | SK-MEL-2 | Mutant (G245S) | Active | [122,149] |
| SK-MEL-28 | Mutant (L145R) | Active | [122,149] | |
| SK-MEL-5 | Wild-type | Active | [122,149] | |
| Malme-3M | Wild-type | Active | [122,150] | |
| MeWo | Mutant (E258K, Q317 c) b | Active | [149] | |
| Leukemia | HL-60 | Null b | Active | [151,152] |
| K-562 | Mutant (Q136fs*13) b | Active | [151,152] | |
| Kasumi-1 | Mutant (R248Q) b | Active | [151] | |
| MOLT-4 | Mutant (R306 c) a | Active | [152] | |
| RPMI-8226 | Mutant (E285K) a | No | [153,154] | |
| CCRF-CEM | Mutant (R248Q, R175H) a | Active | [155] |
| Inhibitor | Target | Mechanism | Phase a | Potential Effects on STAT3/p53 | Refs. |
|---|---|---|---|---|---|
| Napabucasin (BBI-608) | STAT3 | Inhibits gene transcription driven by Stat3 | I/II (Advanced malignancies) III (CRC) | BBI-608 blocks mtp53 (R248Q)-mediated STAT3 activation | [28,167] |
| Celecoxib b | STAT3 | Binds to the three sub-pockets of STAT3 SH2; Inhibits IL-6/STAT3 signaling pathway | III (Breast cancer) II (Lung cancer, Glioblastoma) | Treatment with bortezomib and celecoxib induced apoptosis in p53-degraded cancer cells. Inhibition of COX-2 in colon cancer cell lines by celecoxib increases the nuclear localization of active p53. Celecoxib enhances irradiation-induced apoptosis by p53 signaling. Inhibition of STAT3 pathways by celecoxib induced autophagy, which promoted the degradation of mtp53. | [168,169,170,171,172,173] |
| Ruxolitinib b (INC424) | STAT3 | Targets JAK2/STAT3 axis | IV (Myelofibrosis) II (Leukemia, Lymphoma) | Ruxolitinib blocks mtp53 (R248Q)-mediated STAT3 activation | [28,71] |
| Cetuximab b | STAT3 | Targets EGFR/STAT3 axis | II (Metastatic CRC) I (Squamous Cell Carcinoma, Head and Neck) | The loss of p53 was associated with acquired resistance to EGFR inhibitor. Down-regulation of p53 and up-regulation of EGFR expression increase the sensitivity to cetuximab. Cetuximab administered after oxaliplatin reduces STAT3 phosphorylation and up-regulates p53 protein level. | [174,175,176,177] |
| Sorafenib b | STAT3 | Targets JAK2/STAT3 axis | IV (Advanced hepatocellular carcinoma) III (Renal cell carcinoma; Non small cell lung carcinoma) II (CRC metastatic; Thyroid cancer) | Sorafenib kills cancer cells by activating PUMA, a p53 target and a BH3-only Bcl-2 family protein. Sorafenib has synergistic effects with genistein to increase apoptosis through up-regulation of p53 and p21. | [69,178,179] |
| Sunitinib b | STAT3 | Acts as VEGF, PDGFR inhibitor; inhibits tyrosine kinase phosphorylation of STAT3 | II (Renal carcinoma, pancreatic neuroendocrine tumor metastatic) | Sunitinib treatment increased p53 levels in renal cell carcinoma xenografts Sunitinib induces cellular senescence through p53/DEC1 signaling activation in renal cell carcinoma cells | [70,180,181] |
| KX2-391 | STAT3 | Inhibits SRC-STAT3 axis | II (Bone-metastatic, castration-resistant prostate cancer) I (Lymphoma) | KX2-391 has antiproliferative effect by inducing p53 expression in SRC3T3 and HT29 cells | [182,183] |
| Niclosamide b | STAT3 | Inhibits STAT3 activation, nuclear translocation and transactivation | I (Prostate carcinoma) | Niclosamide can activate p53 function in wild-type cells while reducing the growth of p53-deficient cells and p53 mutant patient-derived ovarian xenografts | [157,158] |
| PRIMA-1Met (APR-246) | p53 | Converts to methylene quinuclidinone which binds to thiol group of mtp53 and restores wtp53 | II (High-grade serous ovarian cancer) I (Hematologic, prostatic neoplasms) | APR-246 exhibited synergistic effect with piperlongumine to induce cell death in mtp53 HNSCC. Piperlongumine has been identified as a potential direct STAT3 inhibitor against breast cancer. | [184,185,186] |
| Ganetespib | p53 | Inhibits HSP90 and degrades mtp53 | II (Lung, colon, rectal cancer, gastrointestinal stromal tumor, breast cancer, melanoma) | Phosphorylation level of STAT3 correlated with HSP90 inhibitor resistance in TNBC cells. Ganatespib inhibited pancreatic cancer cell growth via down-regulation of JAK2-STAT3 pathway. | [187,188,189] |
| Onalespib (AT13387) | p53 | Inhibits HSP90 and degrades mtp53 | I (Advanced malignant solid neoplasm) | AT13387 reduced EGFR/STAT3 in nasopharyngeal carcinoma cells. AT13387 inhibited IL-6-mediated STAT3 phosphorylation in myeloma and breast carcinoma cells. | [190,191,192] |
| Luminespib (AUY922) | p53 | Inhibits HSP90 and degrades mtp53 | II (Non-small cell lung cancer) | NVP-AUY922 enhances TRAIL-induced apoptosis by down-regulating JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells. Treatment with NVP-AUY922 negatively affected IL-6-mediated STAT3 phosphorylation. AUY922 treatment disrupts the association between HSP90 and its client proteins (JAK2, STAT3) and reduced the levels of STAT3. AUY922 treatment inhibited STAT3 activity in chronic lymphocytic leukemia cells. | [193,194,195,196,197] |
| Atorvastatin b | p53 | Inhibits HMG-CoA reductase, disrupts mtp53-HSP90 complex | II (Prostatic neoplasms) | Atorvastatin treatment reduced phosphorylation of STAT3 in head and neck squamous cell carcinoma. The combined use of atorvastatin and aspirin attenuated STAT3 phosphorylation in the treatment of prostate cancer. Atorvastatin induced senescence of hepatocellular carcinoma through downregulation of IL-6/STAT3 pathway. | [198,199,200,201] |
| Lovastatin b | p53 | Inhibits HMG-CoA reductase, disrupts mtp53-HSP90 complex | II (Prostate cancer) | Lovastatin inhibited mevalonate pathway that reduced mtp53 expression, and inhibited STAT3 phosphorylation in glioblastoma and pancreatic cancer cells | [160] |
| Simvastatin b | p53 | Inhibits HMG-CoA reductase, disrupts mtp53-HSP90 complex, degrades mtp53 | III (Gastric cancer) II (Breast cancer) | Simvastatin reduced renal cancer cell growth and metastasis through inhibition of JAK2/STAT3 pathway. Simvastatin induced growth arrest by suppressing STAT3/SKP2 in HCC cells. | [161,202,203] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.-H.; Park, H.-M.; Kim, J.; Hong, J.T.; Yoon, D.-Y. STAT3 and p53: Dual Target for Cancer Therapy. Biomedicines 2020, 8, 637. https://doi.org/10.3390/biomedicines8120637
Pham T-H, Park H-M, Kim J, Hong JT, Yoon D-Y. STAT3 and p53: Dual Target for Cancer Therapy. Biomedicines. 2020; 8(12):637. https://doi.org/10.3390/biomedicines8120637
Chicago/Turabian StylePham, Thu-Huyen, Hyo-Min Park, Jinju Kim, Jin Tae Hong, and Do-Young Yoon. 2020. "STAT3 and p53: Dual Target for Cancer Therapy" Biomedicines 8, no. 12: 637. https://doi.org/10.3390/biomedicines8120637
APA StylePham, T. -H., Park, H. -M., Kim, J., Hong, J. T., & Yoon, D. -Y. (2020). STAT3 and p53: Dual Target for Cancer Therapy. Biomedicines, 8(12), 637. https://doi.org/10.3390/biomedicines8120637