Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases

 ,
,  and
and
Abstract
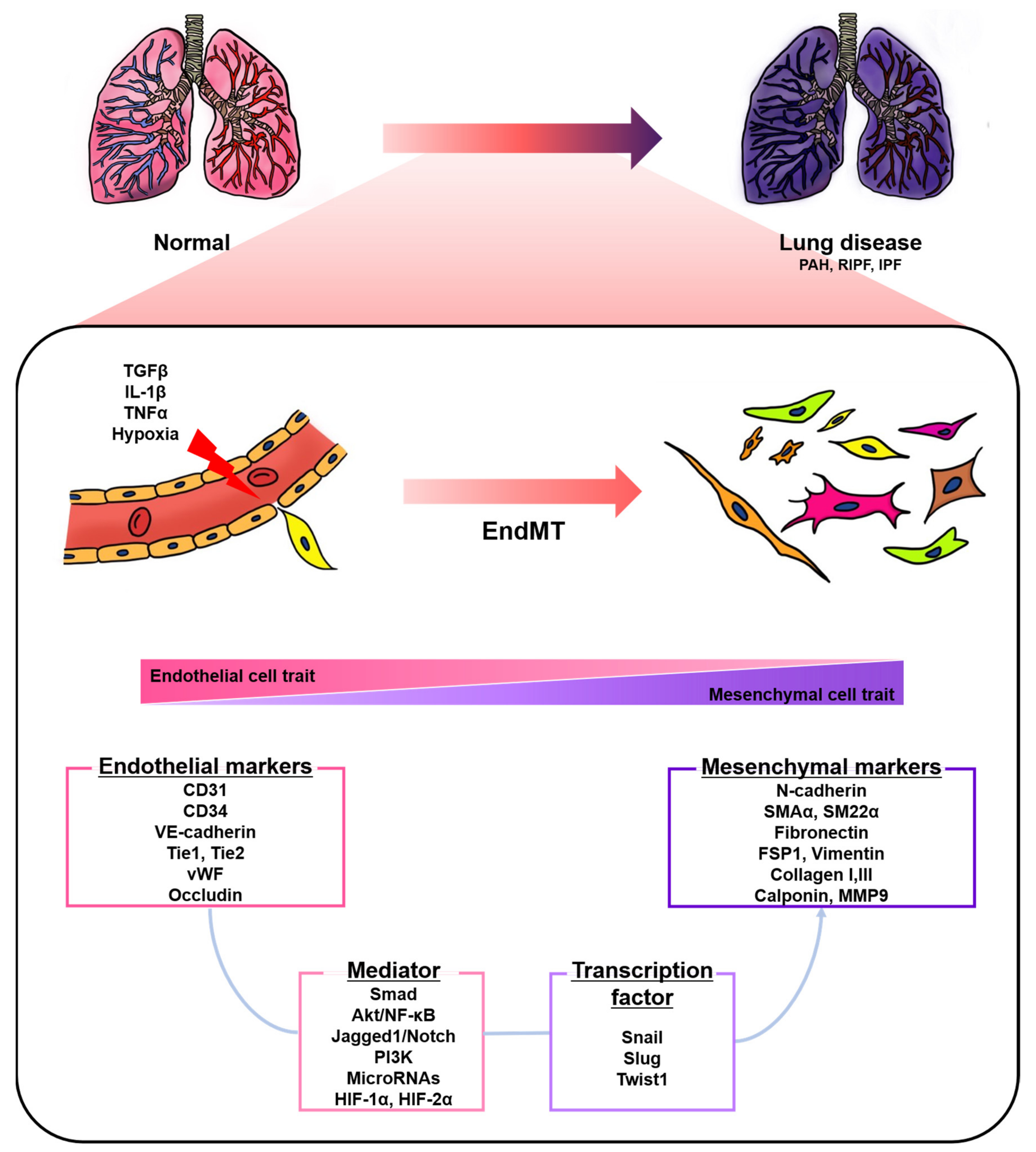
:1. Introduction
2. EndMT in Pulmonary Hypertension
3. EndMT in Pulmonary Fibrosis
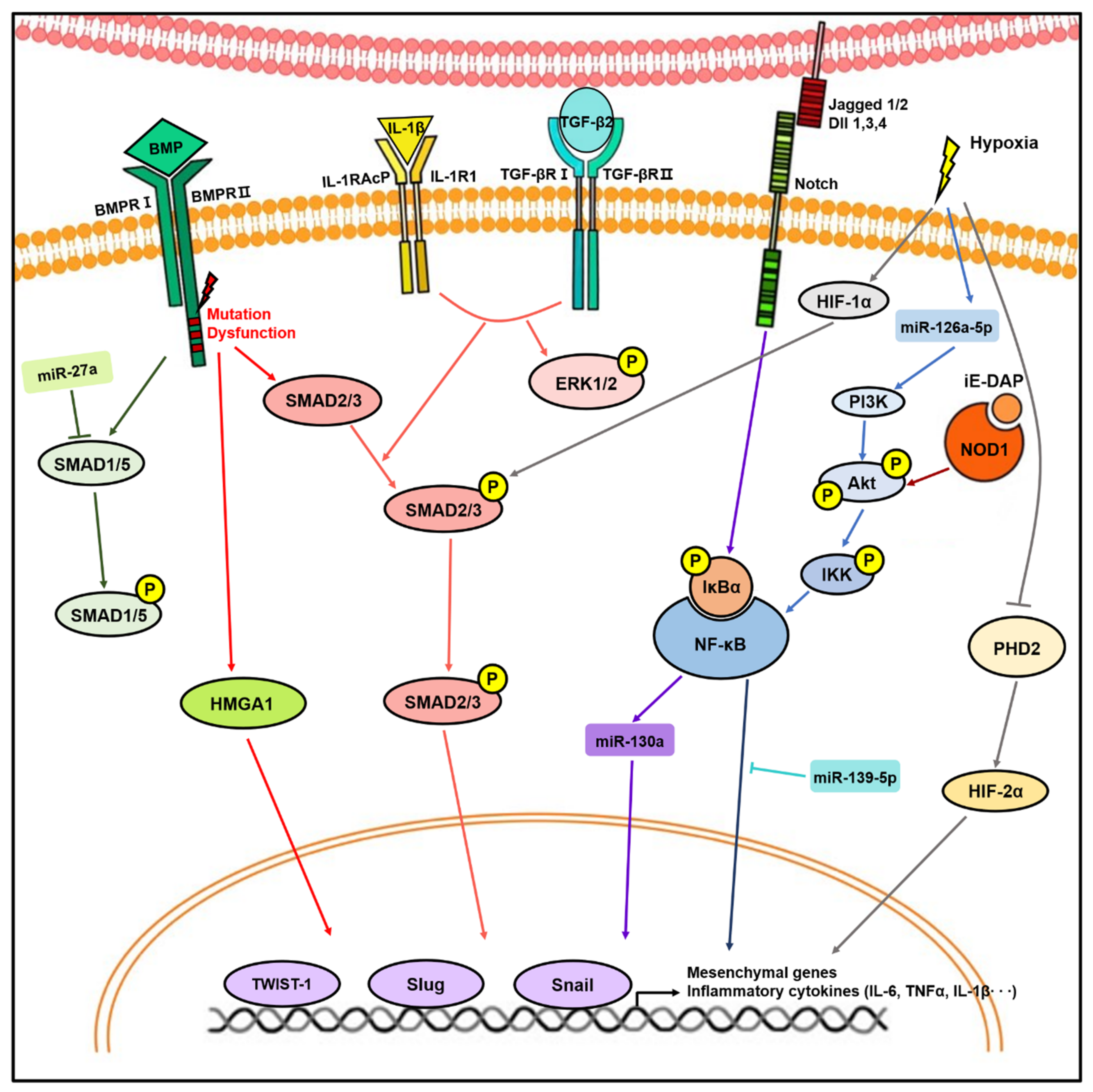
4. Key Signaling Pathways and Mediators during EndMT in Lung Diseases
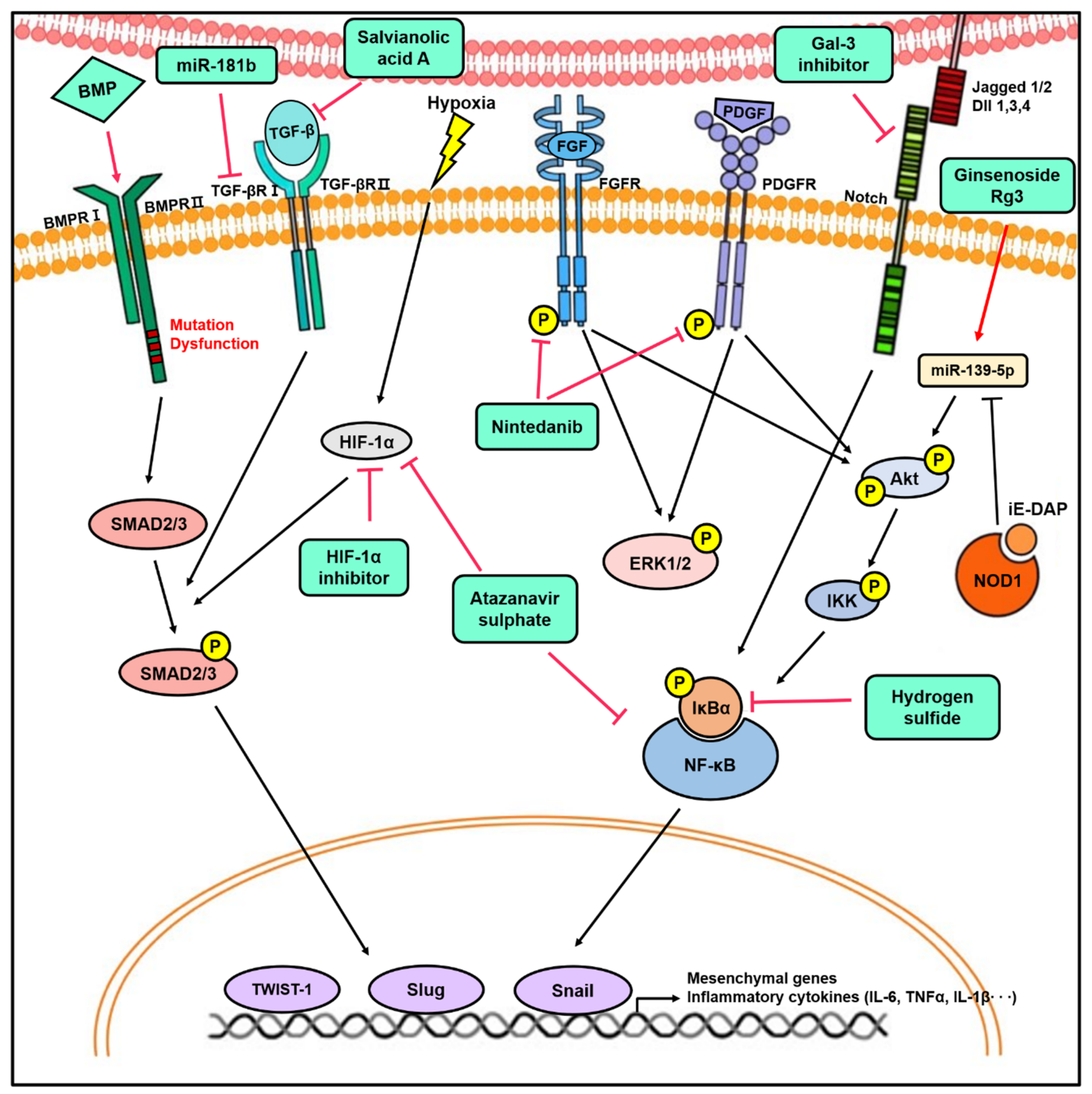
5. Targeting EndMT for Potential Therapeutic Applications
6. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Huh, J.W.; Kim, E.Y.; Shin, M.K.; Park, J.E.; Kim, S.W.; Lee, W.; Choi, B.; Chang, E.J. Endothelial dysfunction induces atherosclerosis: Increased aggrecan expression promotes apoptosis in vascular smooth muscle cells. BMB Rep. 2019, 52, 145–150. [Google Scholar]
- Sena, C.M.; Pereira, A.M.; Seica, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Krenning, G.; Barauna, V.G.; Krieger, J.E.; Harmsen, M.C.; Moonen, J.R. Endothelial plasticity: Shifting phenotypes through force feedback. Stem Cells Int. 2016, 2016, 9762959. [Google Scholar] [CrossRef] [Green Version]
- Sabbineni, H.; Verma, A.; Somanath, P.R. Isoform-specific effects of transforming growth factor beta on endothelial-to-mesenchymal transition. J. Cell. Physiol. 2018, 233, 8418–8428. [Google Scholar] [CrossRef]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thome, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faca, V.; Olsen Camara, N.; Kashima, S.; Tadeu Covas, D. Endothelial cells tissue-specific origins affects their responsiveness to TGF-beta2 during endothelial-to-mesenchymal transition. Int. J. Mol. Sci. 2019, 20, 458. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Guan, S.; Zhou, J. CXCR7 attenuates the TGF-beta-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. Biosyst. 2017, 13, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Thuan, D.T.B.; Zayed, H.; Eid, A.H.; Abou-Saleh, H.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front. Immunol. 2018, 9, 1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Kim, J.D.; Lee, A.; Choi, J.; Park, Y.; Kang, H.; Chang, W.; Lee, M.S.; Kim, J. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Exp. Mol. Med. 2015, 47, e175. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; McLean, D.; Choi, J.; Kang, H.; Chang, W.; Kim, J. Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB Rep. 2014, 47, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Roos-Hesselink, J.W.; Zijlstra, F. Pulmonary hypertension, how to diagnose and who to treat? Neth. Heart J. 2011, 19, 493–494. [Google Scholar] [CrossRef] [Green Version]
- Lyle, M.A.; Davis, J.P.; Brozovich, F.V. Regulation of pulmonary vascular smooth muscle contractility in pulmonary arterial hypertension: Implications for therapy. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.G.; Edwards, W.D.; Kay, J.M.; Rich, S.; Kernis, J.; Schloo, B.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, primary pulmonary hypertension registry. Circulation 1989, 80, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. A report of the American College of Cardiology foundation task force on expert consensus documents and the American Heart Association developed in collaboration with the American College of Chest physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Moonen, J.R.A.J.; Diebold, I.; Cao, A.Q.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.X.; Wang, L.L.; Rabinovitch, M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-mesenchymal transition in pulmonary arterial hypertension. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to mesenchymal transition (EndMT) and vascular remodeling in pulmonary hypertension and idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2020, 14, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zou, X.Z.; Huang, N.; Ge, X.Y.; Yao, M.Z.; Liu, H.; Zhang, Z.; Hu, C.P. MiR-27a promotes endothelial-mesenchymal transition in hypoxia-induced pulmonary arterial hypertension by suppressing BMP signaling. Life Sci. 2019, 227, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yu, M.; Xu, J.; Cheng, Y.; Li, X.; Wei, G.; Wang, H.; Kong, H.; Xie, W. Glucagon-like peptide-1 (GLP-1) mediates the protective effects of dipeptidyl peptidase IV inhibition on pulmonary hypertension. J. Biomed. Sci. 2019, 26, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wang, Y.; Zhang, X.; Guo, Y.; Wang, X. miR-181b-5p inhibits endothelial-mesenchymal transition in monocrotaline-induced pulmonary arterial hypertension by targeting endocan and TGFBR1. Toxicol. Appl. Pharmacol. 2020, 386, 114827. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Shah, S.J. Pulmonary hypertension. JAMA 2012, 308, 1366–1374. [Google Scholar] [CrossRef]
- Isobe, S.; Kataoka, M.; Endo, J.; Moriyama, H.; Okazaki, S.; Tsuchihashi, K.; Katsumata, Y.; Yamamoto, T.; Shirakawa, K.; Yoshida, N.; et al. Endothelial-mesenchymal transition drives expression of CD44 variant and xCT in pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 2019, 61, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Y.M.; Zeng, X.X.; Wang, X.Y.; Chen, S.K.; Gui, L.X.; Lin, M.J. Galectin-3-mediated transdifferentiation of pulmonary artery endothelial cells contributes to hypoxic pulmonary vascular remodeling. Cell. Physiol. Biochem. 2018, 51, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Nagaoka, T.; Yoshida, T.; Wang, L.; Kuriyama, S.; Suzuki, Y.; Nagata, Y.; Harada, N.; Kodama, Y.; Takahashi, F.; et al. Nintedanib ameliorates experimental pulmonary arterial hypertension via inhibition of endothelial mesenchymal transition and smooth muscle cell proliferation. PLoS ONE 2019, 14, e0214697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Yuan, T.Y.; Zhang, H.F.; Yan, Y.; Wang, D.S.; Fang, L.H.; Lu, Y.; Du, G.H. Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: An insight from a plant polyphenol. Int. J. Biol. Sci. 2017, 13, 1067–1081. [Google Scholar] [CrossRef]
- Reynolds, A.M.; Holmes, M.D.; Danilov, S.M.; Reynolds, P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012, 39, 329–343. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Alveolar epithelial cell disintegrity and subsequent activation a key process in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 119–121. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Resp. Cell. Mol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Ji, Y.; Zhang, G.; Zhang, X.; Li, B.; Li, D.; Jiang, W. Protective effect of atazanavir sulphate against pulmonary fibrosis in vivo and in vitro. Basic Clin. Pharmacol. Toxicol. 2018, 122, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Tada, Y.; Gladson, S.; Nishimura, R.; Shimomura, I.; Karasawa, S.; Tatsumi, K.; West, J. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir. Res. 2017, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Nam, J.K.; Kim, B.Y.; Jang, J.; Jin, Y.B.; Lee, H.J.; Park, S.; Ji, Y.H.; Cho, J.; Lee, Y.J. HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. 2016, 76, 1019–1030. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Gong, J.; Dennery, P.A.; Yao, H. Endothelial-to-mesenchymal transition: Pathogenesis and therapeutic targets for chronic pulmonary and vascular diseases. Biochem. Pharm. 2019, 168, 100–107. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Nishimura, T.; Shi, L.; Sessions, D.; Thrasher, A.; Trudell, J.R.; Berry, G.J.; Pearl, R.G.; Kao, P.N. Endothelial fate mapping in mice with pulmonary hypertension. Circulation 2014, 129, 692–703. [Google Scholar] [CrossRef] [Green Version]
- Sakao, S.; Hao, H.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Tatsumi, K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: crosstalk with myofibroblast-like cells. Respir. Res. 2011, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, Y.; Ma, Y.; Zhang, J.; Wang, C.; Zhang, H. Protective effect of hydrogen sulfide on monocrotalineinduced pulmonary arterial hypertension via inhibition of the endothelial mesenchymal transition. Int. J. Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.J.; He, M.Y.; Han, H.H.; Xie, W.P.; Wang, H.; Kong, H. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab Invest. 2018, 98, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Sabbineni, H.; Verma, A.; Artham, S.; Anderson, D.; Amaka, O.; Liu, F.; Narayanan, S.P.; Somanath, P.R. Pharmacological inhibition of beta-catenin prevents EndMT in vitro and vascular remodeling in vivo resulting from endothelial Akt1 suppression. Biochem. Pharmacol. 2019, 164, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Carrier, E.J.; Talati, M.H.; Rathinasabapathy, A.; Chen, X.; Nishimura, R.; Tada, Y.; Tatsumi, K.; West, J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L118–L126. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, D.; Ernst, A.; Kalluri, R. Idiopathic Pulmonary Fibrosis is Associated with Endothelial to Mesenchymal Transition. Am. J. Resp. Cell Mol. 2010, 43, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, P.; Duan, J.X.; Liu, T.; Guan, X.X.; Mei, W.X.; Liu, Y.P.; Sun, G.Y.; Wan, L.; Zhong, W.J.; et al. Aucubin Alleviates Bleomycin-Induced Pulmonary Fibrosis in a Mouse Model. Inflammation 2017, 40, 2062–2073. [Google Scholar] [CrossRef]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 2011, 17, 355–361. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Tada, Y.; Nishimura, R.; Kawasaki, T.; Sekine, A.; Urushibara, T.; Kato, F.; Kinoshita, T.; Ikari, J.; West, J.; et al. Endothelial-to-mesenchymal transition in lipopolysaccharide-induced acute lung injury drives a progenitor cell-like phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1185–L1198. [Google Scholar] [CrossRef] [Green Version]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am. J. Pathol. 2013, 182, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.J.; Chen, I.C.; Li, H.H.; Huang, C.C. EP4 Agonist L-902,688 Suppresses EndMT and Attenuates Right Ventricular Cardiac Fibrosis in Experimental Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-beta-Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Park, S.Y.; Chang, H.R.; Jung, E.Y.; Munkhjargal, A.; Lim, J.S.; Lee, M.S.; Kim, Y. Clinical significance linked to functional defects in bone morphogenetic protein type 2 receptor, BMPR2. BMB Rep. 2017, 50, 308–317. [Google Scholar] [CrossRef]
- Mathew, R.; Huang, J.; Iacobas, S.; Iacobas, D.A. Pulmonary Hypertension Remodels the Genomic Fabrics of Major Functional Pathways. Genes 2020, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.T.; Covas, D.T.; Kashima, S.; Rodrigues, C.O. Endothelial Mesenchymal Transition: Comparative Analysis of Different Induction Methods. Biol. Proced. Online 2016, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Wang, W.; Cui, G.; Yan, L.; Zhang, S. Potential role of the Jagged1/Notch1 signaling pathway in the endothelial-myofibroblast transition during BLM-induced pulmonary fibrosis. J. Cell. Physiol. 2018, 233, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, P.; Bledsoe, G.; Yang, Z.R.; Chao, L.; Chao, J. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp. Cell Res. 2015, 337, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Yun, E.; Chang, W.; Kim, J. Ginsenoside Rg3 protects against iE-DAP-induced endothelial-to-mesenchymal transition by regulating the miR-139-5p-NF-kappaB axis. J. Ginseng Res. 2020, 44, 300–307. [Google Scholar] [CrossRef]
- Kim, J. MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. BMB Rep. 2018, 51, 65–72. [Google Scholar] [CrossRef]
- Salvi, V.; Gianello, V.; Tiberio, L.; Sozzani, S.; Bosisio, D. Cytokine Targeting by miRNAs in Autoimmune Diseases. Front. Immunol. 2019, 10, 15. [Google Scholar] [CrossRef]
- Parikh, V.N.; Jin, R.C.; Rabello, S.; Gulbahce, N.; White, K.; Hale, A.; Cottrill, K.A.; Shaik, R.S.; Waxman, A.B.; Zhang, Y.Y.; et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012, 125, 1520–1532. [Google Scholar] [CrossRef]
- Li, L.; Kim, I.K.; Chiasson, V.; Chatterjee, P.; Gupta, S. NF-kappaB mediated miR-130a modulation in lung microvascular cell remodeling: Implication in pulmonary hypertension. Exp. Cell Res. 2017, 359, 235–242. [Google Scholar] [CrossRef]
- Xu, Y.P.; He, Q.; Shen, Z.; Shu, X.L.; Wang, C.H.; Zhu, J.J.; Shi, L.P.; Du, L.Z. MiR-126a-5p is involved in the hypoxia-induced endothelial-to-mesenchymal transition of neonatal pulmonary hypertension. Hypertens Res 2017, 40, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.Y.; Park, K.K.; Green, D.E.; Bijli, K.M.; Searles, C.D.; Sutliff, R.L.; Hart, C.M. Hypoxia mediates mutual repression between microRNA-27a and PPARgamma in the pulmonary vasculature. PLoS ONE 2013, 8, e79503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Liu, B.; Tang, Y.; Li, F.; Qin, W.; Yuan, X. Irradiated Human Umbilical Vein Endothelial Cells Undergo Endothelial-Mesenchymal Transition via the Snail/miR-199a-5p Axis to Promote the Differentiation of Fibroblasts into Myofibroblasts. BioMed Res. Int. 2018, 2018, 4135806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs. Genes 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Lung Diseases | In Vivo Model | In Vitro Model | Cell Line | Endothelial Markers | Mesenchymal Markers | EndMT Mediators | Reference |
|---|---|---|---|---|---|---|---|
| PH | SuHx, MCT rats | BMPR2 deficiency | PAECs | CD31, VE-cad, CD34, Tie2 | SMAα, Vimentin, p-Vimentin | Twist1 | [7] |
| PH | BMPR2 KO mice | BMPR2 deficiency | EAhy926 | VE-cad | N-cad | Slug, Twist | [11] |
| PH | EC-specific BMPR2 KO mice | BMPR2 deficiency | PAECs | vWF, CD31, VE-cad | SM22α, SMAα, p-Vimentin | HMGA1, Snail, Slug | [23] |
| PH | Hypoxia-exposed rats | Hypoxia | Rat PMVECs | CD31 | SMAα, Collagen I, III | HIF-1α, Twist1 | [27] |
| PH | SuHx mice | Combination of TGFβ1, TNFα, and IL-1β | PAECs | vWF, CD31, VE-cad, Occudin | SMAα, Calponin, Collagen I | Inflammatory cytokines | [30] |
| PH | VE-cad Cre or Tie2 Cre-mTomato/mGFP lineage tracing mice | vWF, CD31, VE-cad, Tie2 | SMAα | Not determined | [48] | ||
| PH | MCT rats, EC-specific phd2, egln1, KO mice | Hypoxia, TGFβ1 | PVECs | CD31, VE-cad | SM22α, Vimentin, FN, SMAα, FSP1 | HIF-2α, Snail, Slug, PHD2 | [50] |
| PF | Radiation-exposed mice | Radiation | PAECs, PMVECs | CD31, VE-cad | SMAα, Vimentin, FSP1, Collagen, MMP9 | TGFβ-RI, Smad2/3, HIF-1α, Snail | [41] |
| PF | Bleomycin-treated Tie2-Cre/CAG-CAT-Lac mice | Combination of Ras and TGFβ | MS-1 | CD31, VE-cad, CD34, Tie2, | SMAα, FN, Collagen I | Not determined | [42] |
| PF | Radiation-exposed mice | HSPB1 deficiency | PAECs, PMVECs | CD31, VE-cad | SMAα | Inflammatory cytokines | [45] |
| Clinical Relevance | In Vitro Model | In Vivo Model | Negative Regulator of EndMT | Reference |
|---|---|---|---|---|
| PH | MCT rat | Rapamycin | [7] | |
| PH | Combination of TGFβ1, TNFα, and IL-1β-treated rat PAECs | MCT rat | miR-181b | [29] |
| PH | Combination of TGFβ1, TNFα, and IL-1β-treated PAECs | SuHx mice | Sulfasalazine | [32] |
| PH | Hypoxia-exposed rat | Galectin-3 inhibitor | [33] | |
| PH | Combination of TGFβ2, TNFα and IL-1β-treated PMVECs | SuHx rat | Nintedanib | [34] |
| PH | TGFβ1-treated PAECs | MCT rat | Salvianolic acid A | [35] |
| PH | TGFβ1-treated PAECs | MCT rat | Hydrogen sulfide | [51] |
| PH | MCT rat | Sitagliptin | [52] | |
| PH | TGFβ-treated HUVECs | MCT rat | EP4 agonist | [64] |
| PH | TGFβ1-treated PMVECs | MCT rat, Hypoxia-exposed rat | BMPR2, rhBMP2, rhBMP7 | [36] |
| PF | Radiation-exposed PAECs | Radiation-exposed mice | HIF-1α inhibitor | [41] |
| PF | CoCl2-treated PMVECs | Bleomycin-treated rat | Atazanavir sulphate | [43] |
| PF | LPS-treated PMVECs | LPS-treated mice | Vildagliptin | [44] |
| PF | Radiation-exposed PMVECs | Radiation-exposed EC conditionally overexpressed HSPB1 mice | HSPB1 | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, E.; Kook, Y.; Yoo, K.H.; Kim, K.I.; Lee, M.-S.; Kim, J.; Lee, A. Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines 2020, 8, 639. https://doi.org/10.3390/biomedicines8120639
Yun E, Kook Y, Yoo KH, Kim KI, Lee M-S, Kim J, Lee A. Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines. 2020; 8(12):639. https://doi.org/10.3390/biomedicines8120639
Chicago/Turabian StyleYun, Eunsik, Yunjin Kook, Kyung Hyun Yoo, Keun Il Kim, Myeong-Sok Lee, Jongmin Kim, and Aram Lee. 2020. "Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases" Biomedicines 8, no. 12: 639. https://doi.org/10.3390/biomedicines8120639
APA StyleYun, E., Kook, Y., Yoo, K. H., Kim, K. I., Lee, M. -S., Kim, J., & Lee, A. (2020). Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines, 8(12), 639. https://doi.org/10.3390/biomedicines8120639