The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases

 ,
, 
 ,
,  and
and
Abstract
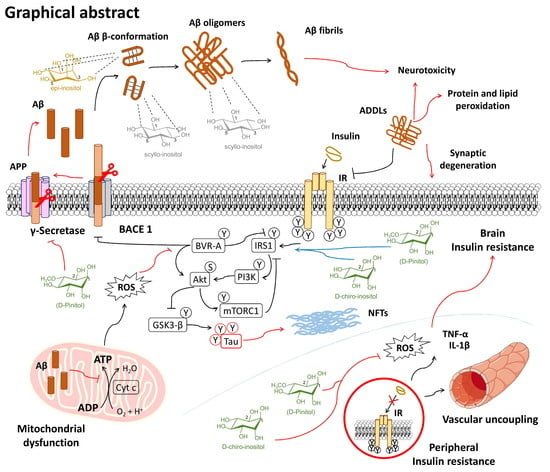
:
1. Introduction: Human Brain Aging and Inositols
2. Inositols in Organisms
2.1. Structure of Inositol Isomers and Inositol Phospholipids
2.2. Inositol Incorporation in Organisms
3. Insulin-Mimetic and Insulin-Sensitizing Properties of Inositols
3.1. Revisiting the Proposed Role of Inositols in Insulin Signaling
3.1.1. Canonical Insulin Signaling
3.1.2. Non-Canonical Insulin Signaling and the Role of IPGs
3.2. Are Inositols Direct Insulin Mimetics Rather than Insulin Sensitizers?
3.3. Putative Role of Inositols in IGF-1 Signaling
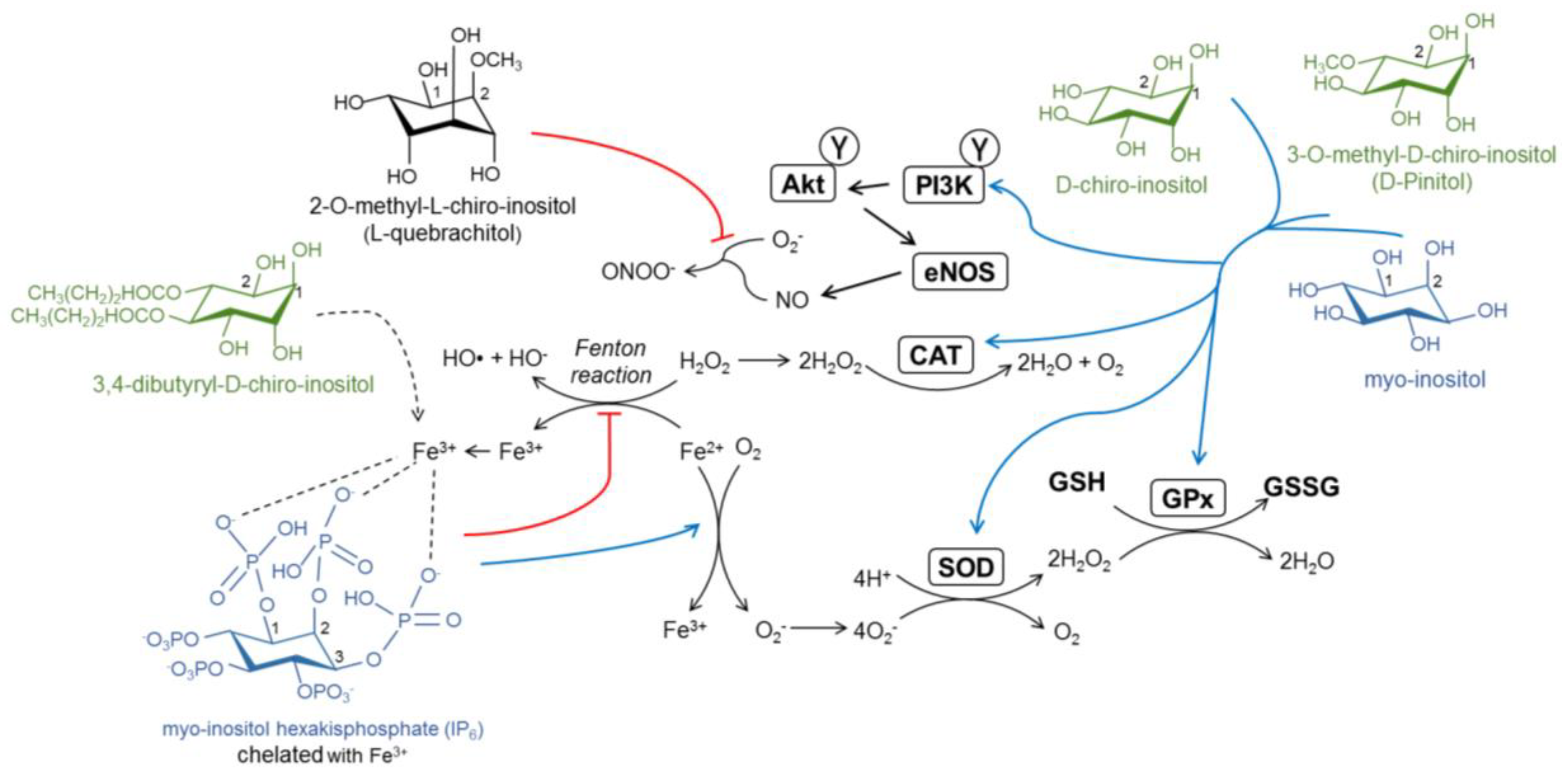
4. The Antioxidant Capacity of Inositols
5. Inositols in the Brain
5.1. Sources and Distribution of Inositols in the Brain
5.2. Inositols as Osmolytes in Astrocytes
5.3. Changes in Inositol Derivatives and Excitability in Neurons
6. Neurodegenerative Diseases: Perspectives for the Use of Inositols
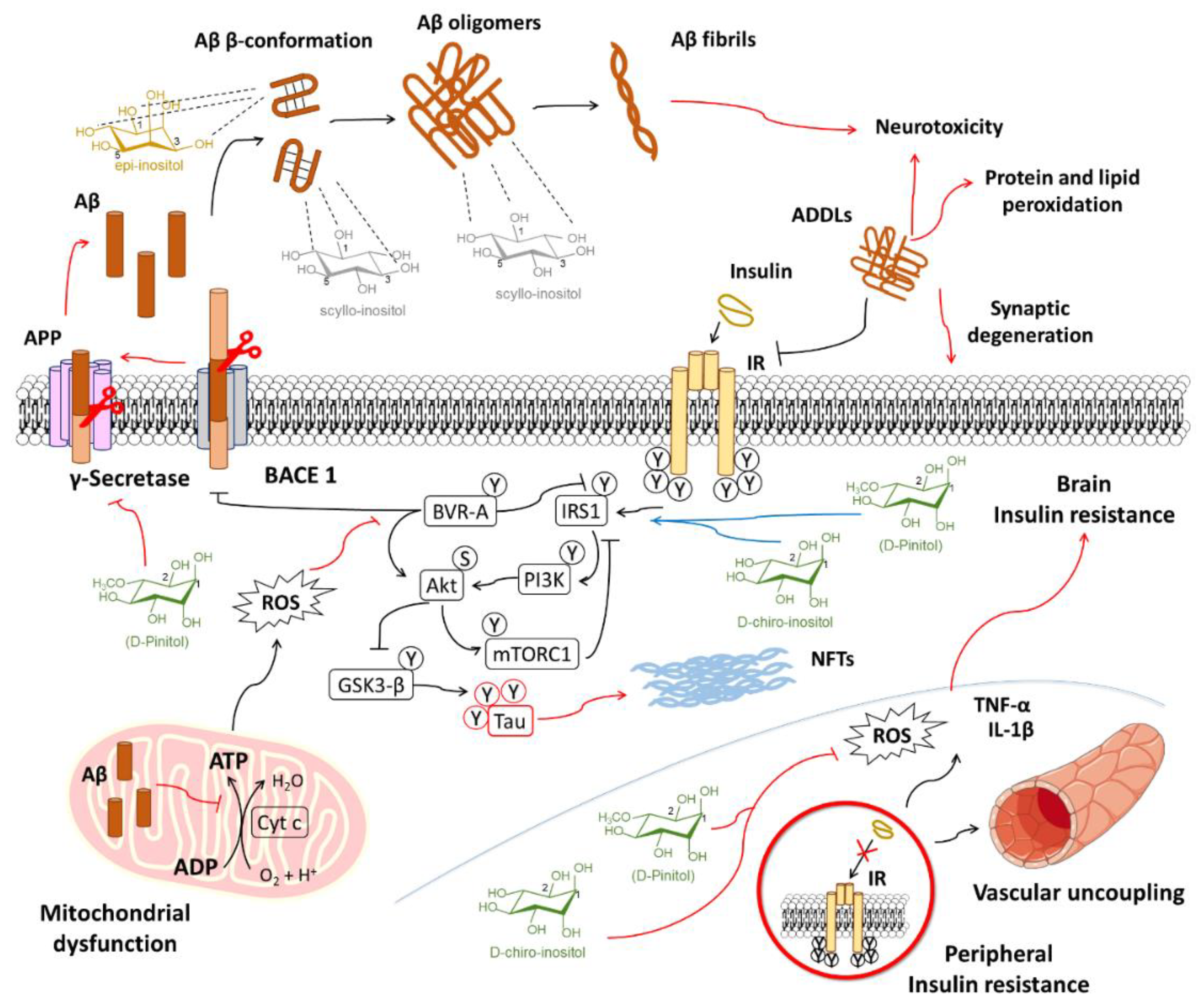
6.1. Alzheimer’s Disease
6.1.1. Inositols and the Amyloid Pathology
6.1.2. Inositol Use for Brain Insulin Resistance in Alzheimer’s Disease
6.1.3. Unhealthy Dietary Habits and Microvascular Damage in Alzheimer’s Disease: Preventive Inositol Supplementation
6.2. Down’s Syndrome
6.3. Anxiety, Compulsive, and Depressive Disorders
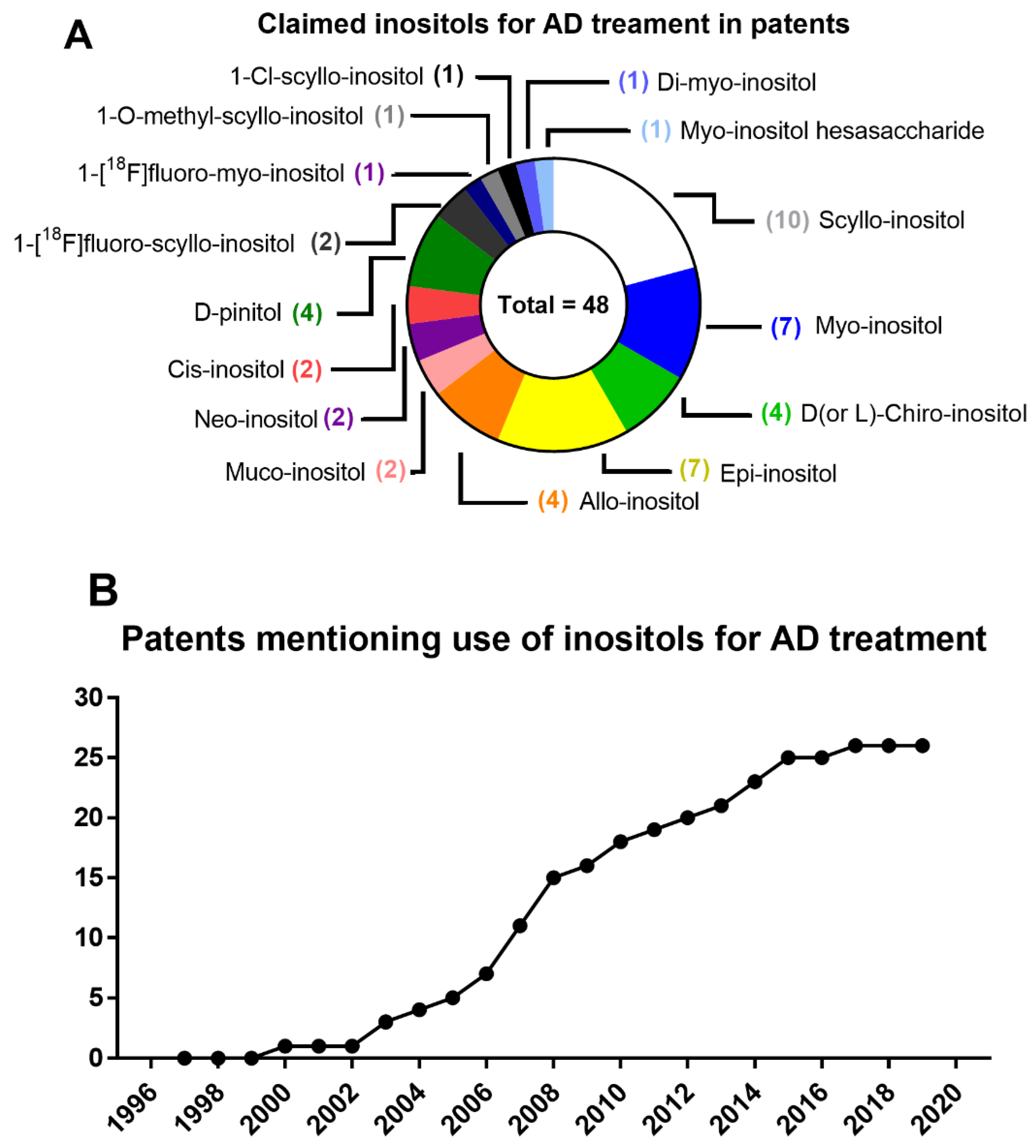
7. From Patents to Clinical Trials of Inositols
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Azzu, V.; Valencak, T.G. Energy metabolism and ageing in the mouse: A mini-review. Gerontology 2017, 63, 327–336. [Google Scholar] [CrossRef]
- Surguchov, A. Caveolin: A new link between diabetes and ad. Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and alzheimer’s disease. Mediat. Inflamm. 2015, 105828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askarova, S.; Umbayev, B.; Masoud, A.-R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The links between the gut microbiome, aging, modern lifestyle and alzheimer’s disease. Front. Cell Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.P.; Mills, S.J.; Potter, B.V. The "other" inositols and their phosphates: Synthesis, biology, and medicine (with Recent Advances in myo-Inositol Chemistry). Angew. Chem. 2016, 55, 1614–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.; Andreassi, A.; Salvioni, M.; Pelliccione, F.; Mantellassi, G.; Banderali, G. Myoinositol and d-chiro inositol in improving insulin resistance in obese male children: Preliminary data. Int. J. Endocrinol. 2016, 8720342. [Google Scholar] [CrossRef] [Green Version]
- Kalra, B.; Kalra, S.; Sharma, J.B. The inositols and polycystic ovary syndrome. Indian J. Endocrinol. Metab. 2016, 20, 720–724. [Google Scholar] [CrossRef]
- Jin, M.; Selkoe, D.J. Systematic analysis of time-dependent neural effects of soluble amyloid β oligomers in culture and in vivo: Prevention by scyllo-inositol. Neurobiol. Dis. 2015, 82, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, D.R. Myo-Inositol and its derivatives: Their emerging role in the treatment of human diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef] [Green Version]
- Larner, J.; Brautigan, D.L.; Thorner, M.O. D-chiro-inositol glycans in insulin signaling and insulin resistance. Mol. Med. 2010, 16, 543–552. [Google Scholar] [CrossRef]
- Owczarczyk-Saczonek, A.; Lahuta, L.B.; Ligor, M.; Placek, W.; Górecki, R.J.; Buszewski, B. The healing-promoting properties of selected cyclitols—A review. Nutrients 2018, 10, 1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipps, P.P.; Holland, W.H.; Sherman, W.R. Interconversion of myo- and scyllo-inositol with simultaneous formation of neo-inositol by an NADP+ dependent epimerase from bovine brain. Biochem. Biophys. Res. Commun. 1977, 77, 340–346. [Google Scholar] [CrossRef]
- Pak, Y.; Huang, L.C.; Lilley, K.J.; Larner, J. In vivo conversion of [3H]myoinositol to [3H]chiroinositol in rat tissues. J. Biol. Chem. 1992, 267, 16904–16910. [Google Scholar] [PubMed]
- Hipps, P.P.; Ackermann, K.E.; Sherman, W.R. Inositol epimerase--inosose reductase from bovine brain. Methods Enzymol. 1982, 89, 593–598. [Google Scholar] [CrossRef]
- Pak, Y.; Paule, C.R.; Bao, Y.D.; Huang, L.C.; Larner, J. Insulin Stimulates The Biosynthesis of Chiro-Inositol-Containing Phospholipids in a Rat Fibroblast Line Expressing the Human Insulin Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 7759–7763. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.H.; Heimark, D.B.; Nguygen, T.; Nadler, J.L.; Larner, J. Both myo-inositol to chiro-inositol epimerase activities and chiro-inositol to myo-inositol ratios are decreased in tissues of GK type 2 diabetic rats compared to Wistar controls. Biochem. Biophys. Res. Commun. 2002, 293, 1092–1098. [Google Scholar] [CrossRef]
- Ryals, P.E.; Kersting, M.C. Sodium-dependent uptake of [3H]scyllo-inositol by Tetrahymena: Incorporation into phosphatidylinositol, phosphatidylinositol-linked glycans, and polyphosphoinositols. Arch. Biochem. Biophys. 1999, 366, 261–266. [Google Scholar] [CrossRef]
- Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J. Properties of scyllo–inositol as a therapeutic treatment of AD-like pathology. J. Mol. Med. 2007, 85, 603–611. [Google Scholar] [CrossRef]
- Santamaria, A.; Di Benedetto, A.; Petrella, E.; Pintaudi, B.; Corrado, F.; D’Anna, R.; Neri, I.; Facchinetti, F. Myo-inositol may prevent gestational diabetes onset in overweight women: A randomized, controlled trial. J. Matern. Fetal Neonatal Med. 2016, 29, 3234–3237. [Google Scholar] [CrossRef]
- Benjamin, J.; Levine, J.; Fux, M.; Aviv, A.; Levy, D.; Belmaker, R.H. Double-blind, placebo-controlled, crossover trial of inositol treatment for panic disorder. J. Clin. Psychopharmacol. 1995, 152, 1084–1086. [Google Scholar] [CrossRef]
- Fux, M.; Levine, J.; Aviv, A.; Belmaker, R.H. Inositol treatment of obsessive-compulsive disorder. Am. J. Psychiatry 1996, 153, 1219–1221. [Google Scholar] [CrossRef] [PubMed]
- Chengappa, K.N.; Levine, J.; Gershon, S.; Mallinger, A.G.; Hardan, A.; Vagnucci, A.; Pollock, B.; Luther, J.; Buttenfield, J.; Verfaille, S.; et al. Inositol as an add-on treatment for bipolar depression. Bipolar Disord. 2000, 2, 47–55. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Franklin, T.; Chakrabartty, A.; Fraser, P.E. Phosphatidylinositol and inositol involvement in Alzheimer amyloid-beta fibril growth and arrest. J. Mol. Biol. 1998, 278, 183–194. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Golomb, R.; Jurewicz, A.; Antel, J.P.; Fraser, P.E. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta -induced toxicity. J. Biol. Chem. 2000, 275, 18495–18502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, L.G.; Schuff, N.; Cashdollar, N.; Weiner, M.W. Scyllo-inositol in normal aging human brain: 1H magnetic resonance spectroscopy study at 4 Tesla. NMR Biomed. 2005, 18, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Stefanis, L. Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J. Neurochem. 2009, 109, 1348–1362. [Google Scholar] [CrossRef]
- Fraticelli, F.; Celentano, C.; Zecca, I.A.; Di Vieste, G.; Pintaudi, B.; Liberati, M.; Franzago, M.; Di Nicola, M.; Vitacolonna, E. Effect of inositol stereoisomers at different dosages in gestational diabetes: An open-label, parallel, randomized controlled trial. Acta Diabetol. 2018, 55, 805–812. [Google Scholar] [CrossRef]
- Nitz, M.; Fenili, D.; Darabie, A.A.; Wu, L.; Cousins, J.E.; McLaurin, J. Modulation of amyloid-beta aggregation and toxicity by inosose stereoisomers. FEBS J. 2008, 275, 1663–1674. [Google Scholar] [CrossRef]
- Dersjant-Li, Y.; Awati, A.; Schulze, H.; Partridge, G. Phytase in non-ruminant animal nutrition: A critical review on phytase activities in the gastrointestinal tract and influencing factors. J. Sci. Food Agric. 2015, 95, 878–896. [Google Scholar] [CrossRef] [Green Version]
- Schlemmer, U.; Jany, K.D.; Berk, A.; Schulz, E.; Rechkemmer, G. Degradation of phytate in the gut of pigs--pathway of gastro-intestinal inositol phosphate hydrolysis and enzymes involved. Arch. Fur. Tierernahr. 2001, 55, 255–280. [Google Scholar] [CrossRef]
- Goodhart, R.S.; Shils, M. Modern Nutrition in Health and Disease; Lea & Febiger: Philadelphia, PA, USA, 1980. [Google Scholar]
- Clements, R.S., Jr.; Darnell, B. Myo-inositol content of common foods: Development of a high-myo-inositol diet. Am. J. Clin. Nutr. 1980, 33, 1954–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, C.-H.; Hossain, M.A.; Lee, E.; Kanth, B.K.; Park, P.B. Increased salt and drought tolerance by D-pinitol production in transgenic Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2018, 504, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Turhan, I. Relationship between sugar profile and d-pinitol content of pods of wild and cultivated types of carob bean (Ceratonia siliqua L.). Int. J. Food Prop. 2014, 17, 363–370. [Google Scholar] [CrossRef]
- Yang, X.W.; Zou, L.; Wu, Q.; Fu, D.X. Studies on chemical constituents from whole plants of Crossostephium chinense. China J. Chin. Mater. Med. 2008, 33, 905–908. [Google Scholar]
- Kim, A.R.; Zou, Y.N.; Park, T.H.; Shim, K.H.; Kim, M.S.; Kim, N.D.; Kim, J.D.; Bae, S.J.; Choi, J.S.; Chung, H.Y. Active components from artemisia iwayomogi displaying ONOO− scavenging activity. Phytother. Res. 2004, 18, 1–7. [Google Scholar] [CrossRef]
- Beemster, P.; Groenen, P.; Steegers-Theunissen, R. Involvement of inositol in reproduction. Nutr. Rev. 2002, 60, 80–87. [Google Scholar] [CrossRef]
- Parthasarathy, R.N.; Lakshmanan, J.; Thangavel, M.; Seelan, R.S.; Stagner, J.I.; Janckila, A.J.; Vadnal, R.E.; Casanova, M.F.; Parthasarathy, L.K. Rat brain myo-inositol 3-phosphate synthase is a phosphoprotein. Mol. Cell. Biochem. 2013, 378, 83–89. [Google Scholar] [CrossRef]
- Yu, W.; Daniel, J.; Mehta, D.; Maddipati, K.R.; Greenberg, M.L. MCK1 is a novel regulator of myo-inositol phosphate synthase (MIPS) that is required for inhibition of inositol synthesis by the mood stabilizer valproate. PLoS ONE 2017, 12, e0182534. [Google Scholar] [CrossRef] [Green Version]
- Dinicola, S.; Minini, M.; Unfer, V.; Verna, R.; Cucina, A.; Bizzarri, M. Nutritional and Acquired Deficiencies in Inositol Bioavailability. Correlations with Metabolic Disorders. Int. J. Mol. Sci. 2017, 18, 2187. [Google Scholar] [CrossRef]
- Foster, D.A.; Salloum, D.; Menon, D.; Frias, M.A. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR). J. Biol. Chem. 2014, 289, 22583–22588. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Ye, C.; Greenberg, M.L. Inositol Hexakisphosphate Kinase 1 (IP6K1) Regulates Inositol Synthesis in Mammalian Cells. J. Biol. Chem. 2016, 291, 10437–10444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, F.; Coady, M.J.; Lapointe, J.Y. Determination of transport stoichiometry for two cation-coupled myo-inositol cotransporters: SMIT2 and HMIT. J. Physiol. 2005, 563, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, R.E., Jr.; Seemayer, R.; Gupta, S.; Kimmel, R.; Ostlund, E.L.; Sherman, W.R. A stereospecific myo-inositol/D-chiro-inositol transporter in HepG2 liver cells. Identification with D-chiro-[3-3H]inositol. J. Biol. Chem. 1996, 271, 10073–10078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, D.A.; Lattimer, S.A. Sodium- and energy-dependent uptake of myo-inositol by rabbit peripheral nerve. Competitive inhibition by glucose and lack of an insulin effect. J. Clin. Investig. 1982, 70, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ma, L.; Fitzgerald, R.L.; Ostlund, R.E., Jr. Human sodium/inositol cotransporter 2 (SMIT2) transports inositols but not glucose in L6 cells. Arch. Biochem. Biophys. 2009, 481, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Fenili, D.; Weng, Y.-Q.; Aubert, I.; Nitz, M.; McLaurin, J. Sodium/myo-Inositol transporters: Substrate transport requirements and regional brain expression in the TgCRND8 mouse model of amyloid pathology. PLoS ONE 2011, 6, e24032. [Google Scholar] [CrossRef] [Green Version]
- Lahjouji, K.; Aouameur, R.; Bissonnette, P.; Coady, M.J.; Bichet, D.G.; Lapointe, J.Y. Expression and functionality of the Na+/myo-inositol cotransporter SMIT2 in rabbit kidney. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Bissonnette, P.; Coady, M.J.; Lapointe, J.Y. Expression of the sodium-myo-inositol cotransporter SMIT2 at the apical membrane of Madin-Darby canine kidney cells. J. Physiol. 2004, 558, 759–768. [Google Scholar] [CrossRef]
- Ostlund, R.E., Jr.; McGill, J.B.; Herskowitz, I.; Kipnis, D.M.; Santiago, J.V.; Sherman, W.R. D-chiro-inositol Metabolism in Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 1993, 90, 9988–9992. [Google Scholar] [CrossRef] [Green Version]
- Asplin, I.; Galasko, G.; Larner, J. Chiro-inositol deficiency and insulin resistance: A comparison of the chiro-inositol- and the myo-inositol-containing insulin mediators isolated from urine, hemodialysate, and muscle of control and type ii diabetic subjects. Proc. Natl. Acad. Sci. USA 1993, 90, 5924–5928. [Google Scholar] [CrossRef] [Green Version]
- Uldry, M.; Ibberson, M.; Horisberger, J.D.; Chatton, J.Y.; Riederer, B.M.; Thorens, B. Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001, 20, 4467–4477. [Google Scholar] [CrossRef] [PubMed]
- Uldry, M.; Steiner, P.; Zurich, M.G.; Beguin, P.; Hirling, H.; Dolci, W.; Thorens, B. Regulated exocytosis of an H+/myo-inositol symporter at synapses and growth cones. EMBO J. 2004, 23, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Daniel, E.; Mok, M.H.S.; Mead, E.; Mutinelli, C.; Zambello, E.; Caberlotto, L.L.; Pell, T.J.; Langmead, C.J.; Shah, A.J.; Duddy, G.; et al. Evaluation of expression and function of the H+/myo-inositol transporter HMIT. BMC Cell Biol. 2009, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Li, B.; Hertz, L.; Peng, L. Contributions in astrocytes of SMIT1/2 and HMIT to myo-inositol uptake at different concentrations and pH. Neurochem. Int. 2012, 61, 187–194. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Michell, R.H. Do inositol supplements enhance phosphatidylinositol supply and thus support endoplasmic reticulum function? Br. J. Nutr. 2018, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Abel, K.; Anderson, R.A.; Shears, S.B. Phosphatidylinositol and inositol phosphate metabolism. J. Cell Sci. 2001, 114, 2207–2208. [Google Scholar]
- Muller, G. Dynamics of plasma membrane microdomains and cross-talk to the insulin signalling cascade. FEBS Lett. 2002, 531, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Schulz, A.; Wied, S.; Frick, W. Regulation of lipid raft proteins by glimepiride- and insulin-induced glycosylphosphatidylinositol-specific phospholipase C in rat adipocytes. Biochem. Pharmacol. 2005, 69, 761–780. [Google Scholar] [CrossRef]
- Croze, M.L.; Soulage, C.O. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 2013, 95, 1811–1827. [Google Scholar] [CrossRef]
- Goel, M.; Azev, V.N.; d’Alarcao, M. The biological activity of structurally defined inositol glycans. Future Med. Chem. 2009, 1, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltiel, A.R.; Pessin, J.E. Insulin signaling pathways in time and space. Trends Cell Biol. 2002, 12, 65–71. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Siegel, M.I.; Jacobs, S.; Cuatrecasas, P. Putative mediators of insulin action: Regulation of pyruvate dehydrogenase and adenylate cyclase activities. Proc. Natl. Acad. Sci. USA 1982, 79, 3513–3517. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Cuatrecasas, P. Insulin Stimulates the Generation From Hepatic Plasma Membranes of Modulators Derived from an Inositol Glycolipid. Proc. Natl. Acad. Sci. USA 1986, 83, 5793–5797. [Google Scholar] [CrossRef] [Green Version]
- Kessler, A.; Muller, G.; Wied, S.; Crecelius, A.; Eckel, J. Signalling pathways of an insulin-mimetic phosphoinositolglycan-peptide in muscle and adipose tissue. Biochem J. 1998, 330 Pt 1, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Kunjara, S.; Wang, D.Y.; Greenbaum, A.L.; McLean, P.; Kurtz, A.; Rademacher, T.W. Inositol phosphoglycans in diabetes and obesity: Urinary levels of IPG A-type and IPG P-type, and relationship to pathophysiological changes. Mol. Genet. Metab. 1999, 68, 488–502. [Google Scholar] [CrossRef]
- Frick, W.; Bauer, A.; Bauer, J.; Wied, S.; Muller, G. Structure-activity relationship of synthetic phosphoinositolglycans mimicking metabolic insulin action. Biochemistry 1998, 37, 13421–13436. [Google Scholar] [CrossRef]
- Larner, J.; Price, J.D.; Heimark, D.; Smith, L.; Rule, G.; Piccariello, T.; Fonteles, M.C.; Pontes, C.; Vale, D.; Huang, L. Isolation, Structure, Synthesis, and Bioactivity of a Novel Putative Insulin Mediator. A Galactosamine chiro-Inositol Pseudo-Disaccharide Mn2+ Chelate with Insulin-like Activity. J. Med. Chem. 2003, 46, 3283–3291. [Google Scholar] [CrossRef]
- Brautigan, D.L.; Brown, M.; Grindrod, S.; Chinigo, G.; Kruszewski, A.; Lukasik, S.M.; Bushweller, J.H.; Horal, M.; Keller, S.; Tamura, S.; et al. Allosteric activation of protein phosphatase 2C by D-chiro-inositol-galactosamine, a putative mediator mimetic of insulin action. Biochemistry 2005, 44, 11067–11073. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, A.; Kikuchi, K.; Tamura, S.; Tsuiki, S. Purification and characterization of Mg2+-dependent glycogen synthase phosphatase (phosphoprotein phosphatase IA) from rat liver. Eur. J. Biochem. 1981, 119, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Maegawa, H.; Egawa, K.; Ugi, S.; Nishio, Y.; Imamura, T.; Kobayashi, T.; Tamura, S.; Olefsky, J.M.; Kashiwagi, A. Protein phosphatase-2C alpha as a positive regulator of insulin sensitivity through direct activation of phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 22715–22726. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Unger, R.H. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E216–E221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, J.B.; Cid, M.B.; Contreras, F.X.; Goni, F.M.; Martin-Lomas, M. Phospholipase cleavage of D- and L-chiro-glycosylphosphoinositides asymmetrically incorporated into liposomal membranes. Chemistry 2006, 12, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Sleight, S.; Wilson, B.A.; Heimark, D.B.; Larner, J. G(q/11) is involved in insulin-stimulated inositol phosphoglycan putative mediator generation in rat liver membranes: Co-localization of G(q/11) with the insulin receptor in membrane vesicles. Biochem. Biophys. Res. Commun. 2002, 295, 561–569. [Google Scholar] [CrossRef]
- Turner, D.I.; Chakraborty, N.; d’Alarcao, M. A fluorescent inositol phosphate glycan stimulates lipogenesis in rat adipocytes by extracellular activation alone. Bioorganic Med. Chem. Lett. 2005, 15, 2023–2025. [Google Scholar] [CrossRef]
- Larner, J. D-chiro-inositol--its functional role in insulin action and its deficit in insulin resistance. Int. J. Exp. Diabetes Res. 2002, 3, 47–60. [Google Scholar] [CrossRef]
- Romero, G.; Gamez, G.; Huang, L.C.; Lilley, K.; Luttrell, L. Anti-inositolglycan antibodies selectively block some of the actions of insulin in intact BC3H1 cells. Proc. Natl. Acad. Sci. USA 1990, 87, 1476–1480. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.F.; Sánchez-Arias, J.A.; Guadaño, A.; Estévez, F.; Varela, I.; Felíu, J.E.; Mato, J.M. Transport in isolated rat hepatocytes of the phospho-oligosaccharide that mimics insulin action. Effects of adrenalectomy and glucocorticoid treatment. Biochem J. 1991, 274 Pt 2, 369–374. [Google Scholar] [CrossRef]
- Suzuki, S.; Sugawara, K.; Satoh, Y.; Toyota, T. Insulin stimulates the generation of two putative insulin mediators, inositol-glycan and diacylglycerol in BC3H-1 myocytes. J. Biol. Chem. 1991, 266, 8115–8121. [Google Scholar] [PubMed]
- Kristiansen, S.; Richter, E.A. GLUT4-containing vesicles are released from membranes by phospholipase D cleavage of a GPI anchor. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E374–E382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Albusac, J.M.; Velazquez, E.; Iglesias, J.; Jimenez, E.; Blazquez, E. Insulin promotes the hydrolysis of a glycosyl phosphatidylinositol in cultured rat astroglial cells. J. Neurochem. 1997, 68, 10–19. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, R.C.; Caldwell, M.; Guo, Y.; Metz, C.; Davitz, M.A.; Olson, L.K.; Deeg, M.A. Mouse glycosylphosphatidylinositol-specific phospholipase D (Gpld1) characterization. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 1998, 9, 710–714. [Google Scholar] [CrossRef]
- Von Toerne, C.; Huth, C.; de Las Heras Gala, T.; Kronenberg, F.; Herder, C.; Koenig, W.; Meisinger, C.; Rathmann, W.; Waldenberger, M.; Roden, M.; et al. MASP1, THBS1, GPLD1 and ApoA-IV are novel biomarkers associated with prediabetes: The KORA F4 study. Diabetologia 2016, 59, 1882–1892. [Google Scholar] [CrossRef]
- Qin, W.; Liang, Y.Z.; Qin, B.Y.; Zhang, J.L.; Xia, N. The Clinical Significance of Glycoprotein Phospholipase D Levels in Distinguishing Early Stage Latent Autoimmune Diabetes in Adults and Type 2 Diabetes. PLoS ONE 2016, 11, e0156959. [Google Scholar] [CrossRef] [Green Version]
- Bowen, R.F.; Raikwar, N.S.; Olson, L.K.; Deeg, M.A. Glucose and insulin regulate glycosylphosphatidylinositol-specific phospholipase D expression in islet beta cells. Metab. Clin. Exp. 2001, 50, 1489–1492. [Google Scholar] [CrossRef]
- Suh, P.G.; Ryu, S.H.; Moon, K.H.; Suh, H.W.; Rhee, S.G. Inositol phospholipid-specific phospholipase C: Complete cDNA and protein sequences and sequence homology to tyrosine kinase-related oncogene products. Proc. Natl. Acad. Sci. USA 1988, 85, 5419–5423. [Google Scholar] [CrossRef] [Green Version]
- Eliakim, R.; Becich, M.J.; Green, K.; Alpers, D.H. Both tissue and serum phospholipases release rat intestinal alkaline phosphatase. Am. J. Physiol. 1990, 259, G618–G625. [Google Scholar] [CrossRef]
- Wu, W.; Wang, L.; Qiu, J.; Li, Z. The analysis of fagopyritols from tartary buckwheat and their anti-diabetic effects in KK-Ay type 2 diabetic mice and HepG2 cells. J. Funct. Foods 2018, 50, 137–146. [Google Scholar] [CrossRef]
- Jones, D.R.; Avila, M.A.; Sanz, C.; Varela-Nieto, I. Glycosyl-phosphatidylinositol-phospholipase type D: A possible candidate for the generation of second messengers. Biochem. Biophys. Res. Commun. 1997, 233, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Suzuki, C.; Hinokio, Y.; Ishigaki, Y.; Katagiri, H.; Kanzaki, M.; Azev, V.N.; Chakraborty, N.; d’Alarcao, M. Insulin-mimicking bioactivities of acylated inositol glycans in several mouse models of diabetes with or without obesity. PLoS ONE 2014, 9, e100466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, K.J.; Hepworth, M.R.; Raikwar, N.S.; Deeg, M.A.; Sevlever, D. Effect of glycosylphosphatidylinositol (GPI)-phospholipase D overexpression on GPI metabolism. Biochem. J. 2004, 378, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, D.L.; O’Brien, K.D.; D’Alessio, D.A.; Brehm, B.J.; Deeg, M.A. Plasma glycosylphosphatidylinositol-specific phospholipase D predicts the change in insulin sensitivity in response to a low-fat but not a low-carbohydrate diet in obese women. Metab. Clin. Exp. 2008, 57, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Deeg, M.A. GPI-specific phospholipase D as an apolipoprotein. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 1994, 27, 375–381. [Google Scholar]
- Deeg, M.A.; Bierman, E.L.; Cheung, M.C. GPI-specific phospholipase D associates with an apoA-I- and apoA-IV-containing complex. J. Lipid Res. 2001, 42, 442–451. [Google Scholar]
- Raikwar, N.S.; Cho, W.K.; Bowen, R.F.; Deeg, M.A. Glycosylphosphatidylinositol-specific phospholipase D influences triglyceride-rich lipoprotein metabolism. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E463–E470. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Vuppalanchi, R.; Raikwar, N.S.; Deeg, M.A. Glycosylphosphatidylinositol-specific phospholipase d in nonalcoholic Fatty liver disease: A preliminary study. J. Clin. Endocrinol. Metab. 2006, 91, 2279–2285. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, R.; Ostadmohammadi, V.; Lankarani, K.B.; Peymani, P.; Akbari, M.; Kolahdooz, F.; Asemi, Z. The effects of inositol supplementation on lipid profiles among patients with metabolic diseases: A systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis. 2018, 17, 123. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.; Nishiumi, S.; Yoshida, K.; Ashida, H. Rat L6 myotubes as an in vitro model system to study GLUT4-dependent glucose uptake stimulated by inositol derivatives. Cytotechnology 2007, 55, 103–108. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, R.; Laviola, L.; Giorgino, F.; Unfer, V.; Bettocchi, S.; Scioscia, M. PKB/Akt and MAPK/ERK phosphorylation is highly induced by inositols: Novel potential insights in endothelial dysfunction in preeclampsia. Pregnancy Hypertens. 2017, 10, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Decara, J.; Medina-Vera, D.; Tovar, R.; Suarez, J.; Pavon, J.; Serrano, A.; Vida, M.; Gutierrez-Adan, A.; Sanjuan, C.; et al. D-Pinitol from Ceratonia siliqua is an orally active natural inositol that reduces pancreas insulin secretion and increases circulating ghrelin levels in Wistar rats. Nutrients 2020, 12, 2030. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shao, M.; Cho, K.W.; Wang, S.; Chen, Z.; Sheng, L.; Wang, T.; Liu, Y.; Rui, L. Herbal constituent sequoyitol improves hyperglycemia and glucose intolerance by targeting hepatocytes, adipocytes, and β-cells. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E932–E940. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [Green Version]
- Yorek, M.A.; Dunlap, J.A.; Thomas, M.J.; Cammarata, P.R.; Zhou, C.; Lowe, W.L., Jr. Effect of TNF-alpha on SMIT mRNA levels and myo-inositol accumulation in cultured endothelial cells. Am. J. Physiol. 1998, 274, C58–C71. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.; Thorner, M.; Brautigan, D.; Larner, J.; Klein, W.L. Protection against the synaptic targeting and toxicity of Alzheimer’s-associated Aβ oligomers by insulin mimetic chiro-inositols. FASEB J. 2013, 27, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.J.; Gandhi, G.R.; Stalin, A.; Balakrishna, K.; Toppo, E.; Sivasankaran, K.; Ignacimuthu, S.; Al-Dhabi, N.A. Myoinositol ameliorates high-fat diet and streptozotocin-induced diabetes in rats through promoting insulin receptor signaling. Biomed. Pharmacother. 2017, 88, 1098–1113. [Google Scholar] [CrossRef]
- Dang, N.T.; Mukai, R.; Yoshida, K.; Ashida, H. D-pinitol and myo-inositol stimulate translocation of glucose transporter 4 in skeletal muscle of C57BL/6 mice. Biosci. Biotechnol. Biochem. 2010, 74, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, M.; Fernandez-Veledo, S.; Vila-Bedmar, R.; Garcia-Guerra, L.; De Alvaro, C.; Nieto-Vazquez, I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J. Anim. Sci. 2008, 86, E94–E104. [Google Scholar] [CrossRef]
- Czauderna, F.; Fechtner, M.; Aygün, H.; Arnold, W.; Klippel, A.; Giese, K.; Kaufmann, J. Functional studies of the PI(3)-kinase signalling pathway employing synthetic and expressed siRNA. Nucleic Acids Res. 2003, 31, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.L.; Park, J.G.; Jiang, Z.Y.; Holik, J.J.; Mitra, P.; Semiz, S.; Guilherme, A.; Powelka, A.M.; Tang, X.; Virbasius, J.; et al. Analysis of insulin signalling by RNAi-based gene silencing. Biochem. Soc. Trans. 2004, 32, 817–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxvig, C. The role of PAPP-A in the IGF system: Location, location, location. J. Cell Commun. Signal. 2015, 9, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farese, R.V.; Nair, G.P.; Standaert, M.L.; Cooper, D.R. Epidermal growth factor and insulin-like growth factor I stimulate the hydrolysis of the insulin-sensitive phosphatidylinositol-glycan in BC3H-1 myocytes. Biochem. Biophys. Res. Commun. 1988, 156, 1346–1352. [Google Scholar] [CrossRef]
- Kojima, I.; Kitaoka, M.; Ogata, E. Insulin-like growth factor-I stimulates diacylglycerol production via multiple pathways in Balb/c 3T3 cells. J. Biol. Chem. 1990, 265, 16846–16850. [Google Scholar]
- Villalba, M.; Alvarez, J.F.; Russell, D.S.; Mato, J.M.; Rosen, O.M. Hydrolysis of glycosyl-phosphatidylinositol in response to insulin is reduced in cells bearing kinase-deficient insulin receptors. Growth Factors 1990, 2, 91–97. [Google Scholar] [CrossRef]
- León, Y.; Vazquez, E.; Sanz, C.; Vega, J.A.; Mato, J.M.; Giraldez, F.; Represa, J.; Varela-Nieto, I. Insulin-like growth factor-I regulates cell proliferation in the developing inner ear, activating glycosyl-phosphatidylinositol hydrolysis and Fos expression. Endocrinology 1995, 136, 3494–3503. [Google Scholar] [CrossRef] [Green Version]
- Romero, G.; Garmey, J.C.; Veldhuis, J.D. The involvement of inositol phosphoglycan mediators in the modulation of steroidogenesis by insulin and insulin-like growth factor-I. Endocrinology 1993, 132, 1561–1568. [Google Scholar] [CrossRef]
- Ruiz-Albusac, J.M.; Zueco, J.A.; Velazquez, E.; Blazquez, E. Insulin does not induce the hydrolysis of a glycosyl phosphatidylinositol in rat fetal hepatocytes. Diabetes 1993, 42, 1262–1272. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [Green Version]
- Maeno, Y.; Li, Q.; Park, K.; Rask-Madsen, C.; Gao, B.; Matsumoto, M.; Liu, Y.; Wu, I.H.; White, M.F.; Feener, E.P.; et al. Inhibition of insulin signaling in endothelial cells by protein kinase C-induced phosphorylation of p85 subunit of phosphatidylinositol 3-kinase (PI3K). J. Biol. Chem. 2012, 287, 4518–4530. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.R.; Crossey, P.A.; Walker, S.; Anilkumar, N.; Poston, L.; Douglas, G.; Ezzat, V.A.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.T. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 2008, 57, 3307–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, E.; Empson, K.L.; Eaton, J.W. Phytic acid. A natural antioxidant. J. Biol. Chem. 1987, 262, 11647–11650. [Google Scholar] [PubMed]
- Graf, E.; Eaton, J.W. Antioxidant functions of phytic acid. Free Radic. Biol. Med. 1990, 8, 61–69. [Google Scholar] [CrossRef]
- Muraoka, S.; Miura, T. Inhibition of xanthine oxidase by phytic acid and its antioxidative action. Life Sci. 2004, 74, 1691–1700. [Google Scholar] [CrossRef]
- Rengarajan, T.; Rajendran, P.; Nandakumar, N.; Balasubramanian, M.P.; Nishigaki, I. Free radical scavenging and antioxidant activity of D-pinitol against 7, 12 dimethylbenz (a) anthracene induced breast cancer in sprague dawley rats. Asian Pac. J. Trop. Dis. 2014, 4, 384–390. [Google Scholar] [CrossRef]
- Nascimento, N.R.F.; Lessa, L.M.A.; Kerntopf, M.R.; Sousa, C.M.; Alves, R.S.; Queiroz, M.G.R.; Price, J.; Heimark, D.B.; Larner, J.; Du, X.; et al. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proc. Natl. Acad. Sci. USA 2006, 103, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.D.; Wu, P.; Kuang, S.Y.; Liu, Y.; Jiang, J.; Hu, K.; Li, S.H.; Tang, L.; Feng, L.; Zhou, X.Q. Myo-inositol prevents copper-induced oxidative damage and changes in antioxidant capacity in various organs and the enterocytes of juvenile Jian carp (Cyprinus carpio var. Jian). Aquat. Toxicol. 2011, 105, 543–551. [Google Scholar] [CrossRef]
- Jiang, W.-D.; Feng, L.; Liu, Y.; Jiang, J.; Zhou, X.-Q. Myo-inositol prevents oxidative damage, inhibits oxygen radical generation and increases antioxidant enzyme activities of juvenile Jian carp (Cyprinus carpio var. Jian). Aquac. Res. 2009, 40, 1770–1776. [Google Scholar] [CrossRef]
- Vasaikar, N.; Mahajan, U.; Patil, K.R.; Suchal, K.; Patil, C.R.; Ojha, S.; Goyal, S.N. D-pinitol attenuates cisplatin-induced nephrotoxicity in rats: Impact on pro-inflammatory cytokines. Chem. Biol. Interact. 2018, 290, 6–11. [Google Scholar] [CrossRef]
- Roman-Ramos, R.; Almanza-Perez, J.C.; Fortis-Barrera, A.; Angeles-Mejia, S.; Banderas-Dorantes, T.R.; Zamilpa-Alvarez, A.; Diaz-Flores, M.; Jasso, I.; Blancas-Flores, G.; Gomez, J.; et al. Antioxidant and anti-inflammatory effects of a hypoglycemic fraction from Cucurbita ficifolia Bouche in streptozotocin-induced diabetes mice. Am. J. Chin. Med. 2012, 40, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.K.; Novak, J.E.; Agranoff, B.W. Inositol and higher inositol phosphates in neural tissues: Homeostasis, metabolism and functional significance. J. Neurochem. 2002, 82, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Kalmbach, S.J.; Hartman, B.K.; Sherman, W.R. Immunohistochemical staining and enzyme activity measurements show myo-inositol-1-phosphate synthase to be localized in the vasculature of brain. J. Neurochem. 1987, 48, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Mejias-Aponte, C.A.; Ye, C.; Bonci, A.; Kiyatkin, E.A.; Morales, M. A subpopulation of neurochemically-identified ventral tegmental area dopamine neurons is excited by intravenous cocaine. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.E.; Turner, R.S.; Agranoff, B.W.; Fisher, S.K. Differentiated human NT2-N neurons possess a high intracellular content of myo-inositol. J. Neurochem. 1999, 72, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, G.T.; Wu, S.; Buccafusca, R.; Ren, J.; Gonzales, L.W.; Ballard, P.L.; Golden, J.A.; Stevens, M.J.; Greer, J.J. Loss of Murine Na+/myo-Inositol Cotransporter Leads to Brain myo-Inositol Depletion and Central Apnea. J. Biol. Chem. 2003, 278, 18297–18302. [Google Scholar] [CrossRef] [Green Version]
- Buccafusca, R.; Venditti, C.P.; Kenyon, L.C.; Johanson, R.A.; Van Bockstaele, E.; Ren, J.; Pagliardini, S.; Minarcik, J.; Golden, J.A.; Coady, M.J.; et al. Characterization of the null murine sodium/myo-inositol cotransporter 1 (Smit1 or Slc5a3) phenotype: Myo-inositol rescue is independent of expression of its cognate mitochondrial ribosomal protein subunit 6 (Mrps6) gene and of phosphatidylinositol levels in neonatal brain. Mol. Genet. Metab. 2008, 95, 81–95. [Google Scholar] [CrossRef]
- Bersudsky, Y.; Shaldubina, A.; Agam, G.; Berry, G.T.; Belmaker, R.H. Homozygote inositol transporter knockout mice show a lithium-like phenotype. Bipolar Disord. 2008, 10, 453–459. [Google Scholar] [CrossRef]
- Cryns, K.; Shamir, A.; Van Acker, N.; Levi, I.; Daneels, G.; Goris, I.; Bouwknecht, J.A.; Andries, L.; Kass, S.; Agam, G.; et al. IMPA1 is essential for embryonic development and lithium-like pilocarpine sensitivity. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Yu, H.; Kruse, M.; Traynor-Kaplan, A.; Hille, B. Osmoregulatory inositol transporter SMIT1 modulates electrical activity by adjusting PI(4,5)P2 levels. Proc. Natl. Acad. Sci. USA 2016, 113, E3290–E3299. [Google Scholar] [CrossRef] [Green Version]
- Dickson, E.J.; Jensen, J.B.; Hille, B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. USA 2014, 111, E2281–E2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, H.; Poitry-Yamate, C.; Preitner, F.; Thorens, B.; Gruetter, R. Neurochemical profile of the mouse hypothalamus using in vivo 1H MRS at 14.1T. NMR Biomed. 2010, 23, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patishi, Y.; Lubrich, B.; Berger, M.; Kofman, O.; van Calker, D.; Belmaker, R.H. Differential uptake of myo-inositol in vivo into rat brain areas. Eur. Neuropsychopharmacol. 1996, 6, 73–75. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Martínez, F.; Salazar, K.; Cifuentes, M.; Nualart, F. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Mol. Neurobiol. 2019, 56, 769–796. [Google Scholar] [CrossRef] [PubMed]
- Raghu, P.; Joseph, A.; Krishnan, H.; Singh, P.; Saha, S. Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Song, S.; Ruz-Maldonado, I.; Pingitore, A.; Huang, G.C.; Baker, D.; Jones, P.M.; Persaud, S.J. GPR55-dependent stimulation of insulin secretion from isolated mouse and human islets of Langerhans. Diabetes Obes. Metab. 2016, 18, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Bjursell, M.; Ryberg, E.; Wu, T.; Greasley, P.J.; Bohlooly-Y., M.; Hjorth, S. Deletion of Gpr55 Results in Subtle Effects on Energy Metabolism, Motor Activity and Thermal Pain Sensation. PLoS ONE 2016, 11, e0167965. [Google Scholar] [CrossRef] [Green Version]
- Deliu, E.; Sperow, M.; Console-Bram, L.; Carter, R.L.; Tilley, D.G.; Kalamarides, D.J.; Kirby, L.G.; Brailoiu, G.C.; Brailoiu, E.; Benamar, K.; et al. The Lysophosphatidylinositol Receptor GPR55 Modulates Pain Perception in the Periaqueductal Gray. Mol. Pharmacol. 2015, 88, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Méndez-Díaz, M.; Prospéro-García, O. Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. 2018, 78, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Marichal-Cancino, B.A.; Sánchez-Fuentes, A.; Méndez-Díaz, M.; Ruiz-Contreras, A.E.; Prospéro-García, O. Blockade of GPR55 in the dorsolateral striatum impairs performance of rats in a T-maze paradigm. Behav. Pharmacol. 2016, 27, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Sylantyev, S.; Jensen, T.P.; Ross, R.A.; Rusakov, D.A. Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc. Natl. Acad. Sci. USA 2013, 110, 5193–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waragai, M.; Moriya, M.; Nojo, T. Decreased N-Acetyl Aspartate/Myo-Inositol Ratio in the Posterior Cingulate Cortex Shown by Magnetic Resonance Spectroscopy May Be One of the Risk Markers of Preclinical Alzheimer’s Disease: A 7-Year Follow-Up Study. J. Alzheimer’s Dis. 2017, 60, 1411–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitolo, M.; Stanzani-Maserati, M.; Capellari, S.; Testa, C.; Rucci, P.; Poda, R.; Oppi, F.; Gallassi, R.; Sambati, L.; Rizzo, G.; et al. Predicting conversion from mild cognitive impairment to Alzheimer’s disease using brain (1)H-MRS and volumetric changes: A two- year retrospective follow-up study. Neuroimage Clin. 2019, 23, 101843. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Alexander, G.E.; Daly, E.M.; Shetty, H.U.; Krasuski, J.S.; Rapoport, S.I.; Schapiro, M.B. High Brain myo-Inositol Levels in the Predementia Phase of Alzheimer’s Disease in Adults With Down’s Syndrome: A 1H MRS Study. Am. J. Psychiatry 1999, 156, 1879–1886. [Google Scholar] [CrossRef]
- Beacher, F.; Simmons, A.; Daly, E.; Prasher, V.; Adams, C.; Margallo-Lana, M.L.; Morris, R.; Lovestone, S.; Murphy, K.; Murphy, D.G.M. Hippocampal Myo-inositol and Cognitive Ability in Adults With Down Syndrome: An In Vivo Proton Magnetic Resonance Spectroscopy Study. Arch. Gen. Psychiatry 2005, 62, 1360–1365. [Google Scholar] [CrossRef] [Green Version]
- Neverisky, D.L.; Abbott, G.W. KCNQ-SMIT complex formation facilitates ion channel-solute transporter cross talk. FASEB J. 2017, 31, 2828–2838. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [Green Version]
- Manville, R.W.; Abbott, G.W. Potassium channels act as chemosensors for solute transporters. Commun. Biol. 2020, 3, 90. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, T.; Rhee, H.J.; Subramanian, D.; Paraskevopoulou, F.; Mueller, R.; Schultz, C.; Brose, N.; Rhee, J.S.; Betz, H. Endosomal Phosphatidylinositol 3-Phosphate Promotes Gephyrin Clustering and GABAergic Neurotransmission at Inhibitory Postsynapses. J. Biol. Chem. 2017, 292, 1160–1177. [Google Scholar] [CrossRef] [Green Version]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, F.; Green, E.M.; Rousset, M.; Dolmetsch, R.E. PIKfyve regulates CaV1.2 degradation and prevents excitotoxic cell death. J. Cell Biol. 2009, 187, 279–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebohm, G.; Neumann, S.; Theiss, C.; Novkovic, T.; Hill, E.V.; Tavaré, J.M.; Lang, F.; Hollmann, M.; Manahan-Vaughan, D.; Strutz-Seebohm, N. Identification of a novel signaling pathway and its relevance for GluA1 recycling. PLoS ONE 2012, 7, e33889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.X.; Duan, L.H.; He, S.J.; Zhuang, G.F.; Yu, X. Phosphatidylinositol 3,4-bisphosphate regulates neurite initiation and dendrite morphogenesis via actin aggregation. Cell Res. 2017, 27, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Richter-Landsberg, C.; Leibfritz, D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev. Neurosci. 1993, 15, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, L.; Strange, K. In situ localization and osmotic regulation of the Na(+)-myo-inositol cotransporter in rat brain. Am. J. Physiol. 1996, 271, F877–F885. [Google Scholar] [CrossRef]
- Jackson, P.S.; Morrison, R.; Strange, K. The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am. J. Physiol. 1994, 267, C1203–C1209. [Google Scholar] [CrossRef]
- Harris, J.L.; Yeh, H.-W.; Choi, I.-Y.; Lee, P.; Berman, N.E.; Swerdlow, R.H.; Craciunas, S.C.; Brooks, W.M. Altered neurochemical profile after traumatic brain injury: (1)H-MRS biomarkers of pathological mechanisms. J. Cereb. Blood Flow Metab. 2012, 32, 2122–2134. [Google Scholar] [CrossRef] [Green Version]
- Cordoba, J.; Gottstein, J.; Blei, A.T. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: Implications for ammonia-induced brain edema. Hepatology 1996, 24, 919–923. [Google Scholar] [CrossRef]
- Filibian, M.; Frasca, A.; Maggioni, D.; Micotti, E.; Vezzani, A.; Ravizza, T. In vivo imaging of glia activation using 1H-magnetic resonance spectroscopy to detect putative biomarkers of tissue epileptogenicity. Epilepsia 2012, 53, 1907–1916. [Google Scholar] [CrossRef]
- Harris, J.L.; Yeh, H.-W.; Swerdlow, R.H.; Choi, I.-Y.; Lee, P.; Brooks, W.M. High-field proton magnetic resonance spectroscopy reveals metabolic effects of normal brain aging. Neurobiol. Aging 2014, 35, 1686–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, H.; Wu, J.; Zhang, X.; Liu, M.; Wang, Y. Metabonomic alterations in hippocampus, temporal and prefrontal cortex with age in rats. Neurochem. Int. 2009, 54, 481–487. [Google Scholar] [CrossRef] [PubMed]
- von Leden, R.E.; Khayrullina, G.; Moritz, K.E.; Byrnes, K.R. Age exacerbates microglial activation, oxidative stress, inflammatory and NOX2 gene expression, and delays functional recovery in a middle-aged rodent model of spinal cord injury. J. NeuroInflamm. 2017, 14, 161. [Google Scholar] [CrossRef] [PubMed]
- Silverstone, P.H.; McGrath, B.M.; Kim, H. Bipolar disorder and myo-inositol: A review of the magnetic resonance spectroscopy findings. Bipolar Disord. 2005, 7, 1–10. [Google Scholar] [CrossRef]
- Shyng, S.L.; Nichols, C.G. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 1998, 282, 1138–1141. [Google Scholar] [CrossRef]
- Baukrowitz, T.; Schulte, U.; Oliver, D.; Herlitze, S.; Krauter, T.; Tucker, S.J.; Ruppersberg, J.P.; Fakler, B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 1998, 282, 1141–1144. [Google Scholar] [CrossRef]
- Ribalet, B.; John, S.A.; Weiss, J.N. Regulation of cloned ATP-sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP(2)): A study of channel rundown and reactivation. J. Gen. Physiol. 2000, 116, 391–410. [Google Scholar] [CrossRef] [Green Version]
- Enkvetchakul, D.; Loussouarn, G.; Makhina, E.; Shyng, S.L.; Nichols, C.G. The kinetic and physical basis of K(ATP) channel gating: Toward a unified molecular understanding. Biophys. J. 2000, 78, 2334–2348. [Google Scholar] [CrossRef] [Green Version]
- Lacin, E.; Aryal, P.; Glaaser, I.W.; Bodhinathan, K.; Tsai, E.; Marsh, N.; Tucker, S.J.; Sansom, M.S.P.; Slesinger, P.A. Dynamic role of the tether helix in PIP(2)-dependent gating of a G protein-gated potassium channel. J. Gen. Physiol. 2017, 149, 799–811. [Google Scholar] [CrossRef]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohacs, T.; Lopes, C.M.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Suh, B.-C.; Hille, B. Electrostatic interaction of internal Mg2+ with membrane PIP2 Seen with KCNQ K+ channels. J. Gen. Physiol. 2007, 130, 241–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manville, R.W.; Neverisky, D.L.; Abbott, G.W. SMIT1 Modifies KCNQ Channel Function and Pharmacology by Physical Interaction with the Pore. Biophys. J. 2017, 113, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Mayordomo-Cava, J.; Yajeya, J.; Navarro-López, J.D.; Jiménez-Díaz, L. Amyloid-β(25-35) Modulates the Expression of GirK and KCNQ Channel Genes in the Hippocampus. PLoS ONE 2015, 10, e0134385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef]
- Maljevic, S.; Vejzovic, S.; Bernhard, M.K.; Bertsche, A.; Weise, S.; Döcker, M.; Lerche, H.; Lemke, J.R.; Merkenschlager, A.; Syrbe, S. Novel KCNQ3 Mutation in a Large Family with Benign Familial Neonatal Epilepsy: A Rare Cause of Neonatal Seizures. Mol. Syndr. 2016, 7, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Judy, J.T.; Seifuddin, F.; Pirooznia, M.; Mahon, P.B.; Bipolar Genome Study, C.; Jancic, D.; Goes, F.S.; Schulze, T.; Cichon, S.; Noethen, M.; et al. Converging Evidence for Epistasis between ANK3 and Potassium Channel Gene KCNQ2 in Bipolar Disorder. Front. Genet. 2013, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Kaminsky, Z.; Jones, I.; Verma, R.; Saleh, L.; Trivedi, H.; Guintivano, J.; Akman, R.; Zandi, P.; Lee, R.S.; Potash, J.B. DNA methylation and expression of KCNQ3 in bipolar disorder. Bipolar Disord. 2015, 17, 150–159. [Google Scholar] [CrossRef]
- Augelli-Szafran, C.E.; Wei, H.X.; Lu, D.; Zhang, J.; Gu, Y.; Yang, T.; Osenkowski, P.; Ye, W.; Wolfe, M.S. Discovery of notch-sparing gamma-secretase inhibitors. Curr. Alzheimer Res. 2010, 7, 207–209. [Google Scholar] [CrossRef]
- Koppaka, V.; Axelsen, P.H. Accelerated accumulation of amyloid beta proteins on oxidatively damaged lipid membranes. Biochemistry 2000, 39, 10011–10016. [Google Scholar] [CrossRef]
- McLaurin, J.; Kierstead, M.E.; Brown, M.E.; Hawkes, C.A.; Lambermon, M.H.; Phinney, A.L.; Darabie, A.A.; Cousins, J.E.; French, J.E.; Lan, M.F.; et al. Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 2006, 12, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Cleary, J.P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O’Hare, E.; Walsh, D.M.; Selkoe, D.J. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann. Neurol. 2006, 60, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Morrone, C.D.; Bazzigaluppi, P.; Beckett, T.L.; Hill, M.E.; Koletar, M.M.; Stefanovic, B.; McLaurin, J. Regional differences in Alzheimer’s disease pathology confound behavioural rescue after amyloid-β attenuation. Brain 2019, 143, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aytan, N.; Choi, J.-K.; Carreras, I.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Combination therapy in a transgenic model of Alzheimer’s disease. Exp. Neurol. 2013, 250, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, A.; Sahota, B.; Chinta, L.V.; Brown, M.E.; Lai, A.Y.; Ma, K.; Hawkes, C.A.; McLaurin, J.; Stefanovic, B. Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 2012, 135, 3039–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Raleigh, D.P. General amyloid inhibitors? A critical examination of the inhibition of IAPP amyloid formation by inositol stereoisomers. PLoS ONE 2014, 9, e104023. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Du, Z.; Maiti, P.; Klärner, F.-G.; Schrader, T.; Wang, C.; Bitan, G. Comparison of three amyloid assembly inhibitors: The sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem. Neurosci. 2012, 3, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Shea, J.E. Effects of solvent on the structure of the Alzheimer amyloid-beta(25-35) peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Bleiholder, C.; Do, T.D.; Wu, C.; Economou, N.J.; Bernstein, S.S.; Buratto, S.K.; Shea, J.E.; Bowers, M.T. Ion mobility spectrometry reveals the mechanism of amyloid formation of Abeta(25-35) and its modulation by inhibitors at the molecular level: Epigallocatechin gallate and scyllo-inositol. J. Am. Chem. Soc. 2013, 135, 16926–16937. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A.P.; van Dyck, C.H.; Tariot, P.N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 2011, 77, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Liang, E.; Garzone, P.; Cedarbaum, J.M.; Koller, M.; Tran, T.; Xu, V.; Ross, B.; Jhee, S.S.; Ereshefsky, L.; Pastrak, A.; et al. Pharmacokinetic Profile of Orally Administered Scyllo-Inositol (Elnd005) in Plasma, Cerebrospinal Fluid and Brain, and Corresponding Effect on Amyloid-Beta in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2013, 2, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, W.-S.; Lim, S.; Kim, Y.K.; Jung, H.-Y.; Das, S.; Lee, J.; Luo, W.; Kim, K.-T.; Chung, S.-K. A guanidine-appended scyllo-inositol derivative AAD-66 enhances brain delivery and ameliorates Alzheimer’s phenotypes. Sci. Rep. 2017, 7, 14125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinetti, G.M. Compositions and Methods for Treating Alzheimer’s Disease and Related Disorders and Promoting A Healthy Nervous System. U.S. Patent 8,921,347, 30 December 2014. [Google Scholar]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase-A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Frey, W.H. Strategies for Intranasal Delivery of Therapeutics for the Prevention and Treatment of NeuroAIDS. J. Neuroimmune Pharmacol. 2007, 2, 81–86. [Google Scholar] [CrossRef]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Santiago, J.C.P.; Hallschmid, M. Outcomes and clinical implications of intranasal insulin administration to the central nervous system. Exp. Neurol. 2019, 317, 180–190. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isoda, F.; Shiry, L.; Abergel, J.; Allan, G.; Mobbs, C. D-chiro-Inositol enhances effects of hypothalamic toxin gold-thioglucose. Brain Res. 2003, 993, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, A.R.; Menduni, M.; Del Toro, R.; Kyanvash, S.; Maggi, D.; Guglielmi, C.; Pantano, A.L.; Defeudis, G.; Fioriti, E.; Manfrini, S.; et al. A pilot study of D-chiro-inositol plus folic acid in overweight patients with type 1 diabetes. Acta Diabetol. 2017, 54, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C. Diabetes as a risk factor for Alzheimer’s disease: Insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 891–897. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.Y.; Wanrooy, B.J.; Bruce, D.G. Chapter 10-Neuroinflammation, Type 2 Diabetes, and Dementia. In Type 2 Diabetes and Dementia; Srikanth, V., Arvanitakis, Z., Eds.; Academic Press: London, UK, 2018; pp. 195–209. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Pallas, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of beta-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Choi, M.-S.; Han, S.N. High fat diet-induced obesity leads to proinflammatory response associated with higher expression of NOD2 protein. Nutr. Res. Pract. 2011, 5, 219–223. [Google Scholar] [CrossRef]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Hoscheidt, S.M.; Kellawan, J.M.; Berman, S.E.; Rivera-Rivera, L.A.; Krause, R.A.; Oh, J.M.; Beeri, M.S.; Rowley, H.A.; Wieben, O.; Carlsson, C.M.; et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle-aged adults. J. Cereb. Blood Flow Metab. 2017, 37, 2249–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, C.F.; Ledo, A.; Barbosa, R.M.; Laranjinha, J. Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp. Neurol. 2017, 291, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, D.R.; Tucker, K.; Cavallin, M.A.; Mast, T.G.; Fadool, D.A. Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J. Neurosci. 2009, 29, 6734–6751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, J.D.; Yanckello, L.M.; Chlipala, G.; Hammond, T.C.; McCulloch, S.D.; Parikh, I.; Sun, S.; Morganti, J.M.; Green, S.J.; Lin, A.L. Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS ONE 2019, 14, e0221828. [Google Scholar] [CrossRef]
- Oliver, C.; Crayton, L.; Holland, A.; Hall, S.; Bradbury, J. A four year prospective study of age-related cognitive change in adults with Down’s syndrome. Psychol. Med. 1998, 28, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, A.M.; Fernandez-Olivares, P.; Diaz-Franulic, I.; Gonzalez-Jamett, A.M.; Shimahara, T.; Segura-Aguilar, J.; Caviedes, R.; Caviedes, P. Knockdown of Myo-Inositol Transporter SMIT1 Normalizes Cholinergic and Glutamatergic Function in an Immortalized Cell Line Established from the Cerebral Cortex of a Trisomy 16 Fetal Mouse, an Animal Model of Human Trisomy 21 (Down Syndrome). Neurotox. Res. 2017, 32, 614–623. [Google Scholar] [CrossRef]
- Rumble, B.; Retallack, R.; Hilbich, C.; Simms, G.; Multhaup, G.; Martins, R.; Hockey, A.; Montgomery, P.; Beyreuther, K.; Masters, C.L. Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. New Engl. J. Med. 1989, 320, 1446–1452. [Google Scholar] [CrossRef]
- Perluigi, M.; Pupo, G.; Tramutola, A.; Cini, C.; Coccia, R.; Barone, E.; Head, E.; Butterfield, D.A.; Di Domenico, F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta 2014, 1842, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F.; Head, E.; Butterfield, D.A.; Perluigi, M.; Barone, E. Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol. Dis. 2020, 137, 104772. [Google Scholar] [CrossRef]
- Rafii, M.S.; Skotko, B.G.; McDonough, M.E.; Pulsifer, M.; Evans, C.; Doran, E.; Muranevici, G.; Kesslak, P.; Abushakra, S.; Lott, I.T.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study of Oral ELND005 (scyllo-Inositol) in Young Adults with Down Syndrome without Dementia. J. Alzheimer’s Dis. 2017, 58, 401–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, R.; Metzler, D.; Fischer, P.; Heiden, A.; Scharfetter, J.; Moser, E.; Kasper, S. Myo-inositol in depressive and healthy subjects determined by frontal 1H-magnetic resonance spectroscopy at 1.5 tesla. J. Psychiatr. Res. 1998, 32, 411–420. [Google Scholar] [CrossRef]
- Urrila, A.S.; Hakkarainen, A.; Castaneda, A.; Paunio, T.; Marttunen, M.; Lundbom, N. Frontal Cortex Myo-Inositol Is Associated with Sleep and Depression in Adolescents: A Proton Magnetic Resonance Spectroscopy Study. Neuropsychobiology 2017, 75, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Neuman, R.S. Myo-inositol reduces serotonin (5-HT2) receptor induced homologous and heterologous desensitization. Brain Res. 1993, 631, 349–351. [Google Scholar] [CrossRef]
- Einat, H.; Clenet, F.; Shaldubina, A.; Belmaker, R.H.; Bourin, M. The antidepressant activity of inositol in the forced swim test involves 5-HT2 receptors. Behav. Brain Res. 2001, 118, 77–83. [Google Scholar] [CrossRef]
- Harvey, B.H.; Scheepers, A.; Brand, L.; Stein, D.J. Chronic inositol increases striatal D(2) receptors but does not modify dexamphetamine-induced motor behavior. Relev. Obs. Compuls. Disord. Pharmacol. Biochem. Behav. 2001, 68, 245–253. [Google Scholar] [CrossRef]
- Leppink, E.W.; Redden, S.A.; Grant, J.E. A double-blind, placebo-controlled study of inositol in trichotillomania. Int. Clin. Psychopharmacol. 2017, 32, 107–114. [Google Scholar] [CrossRef]
- Seedat, S.; Stein, D.J. Inositol augmentation of serotonin reuptake inhibitors in treatment-refractory obsessive-compulsive disorder: An open trial. Int. Clin. Psychopharmacol. 1999, 14, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Grossman, H.; Marzloff, G.; Luo, X.; LeRoith, D.; Sano, M.; Pasinetti, G. P1-279: NIC5-15 as a treatment for Alzheimer’s: Safety, pharmacokinetics and clinical variables. Alzheimer’s Dement. 2009, 5, 259. [Google Scholar] [CrossRef]






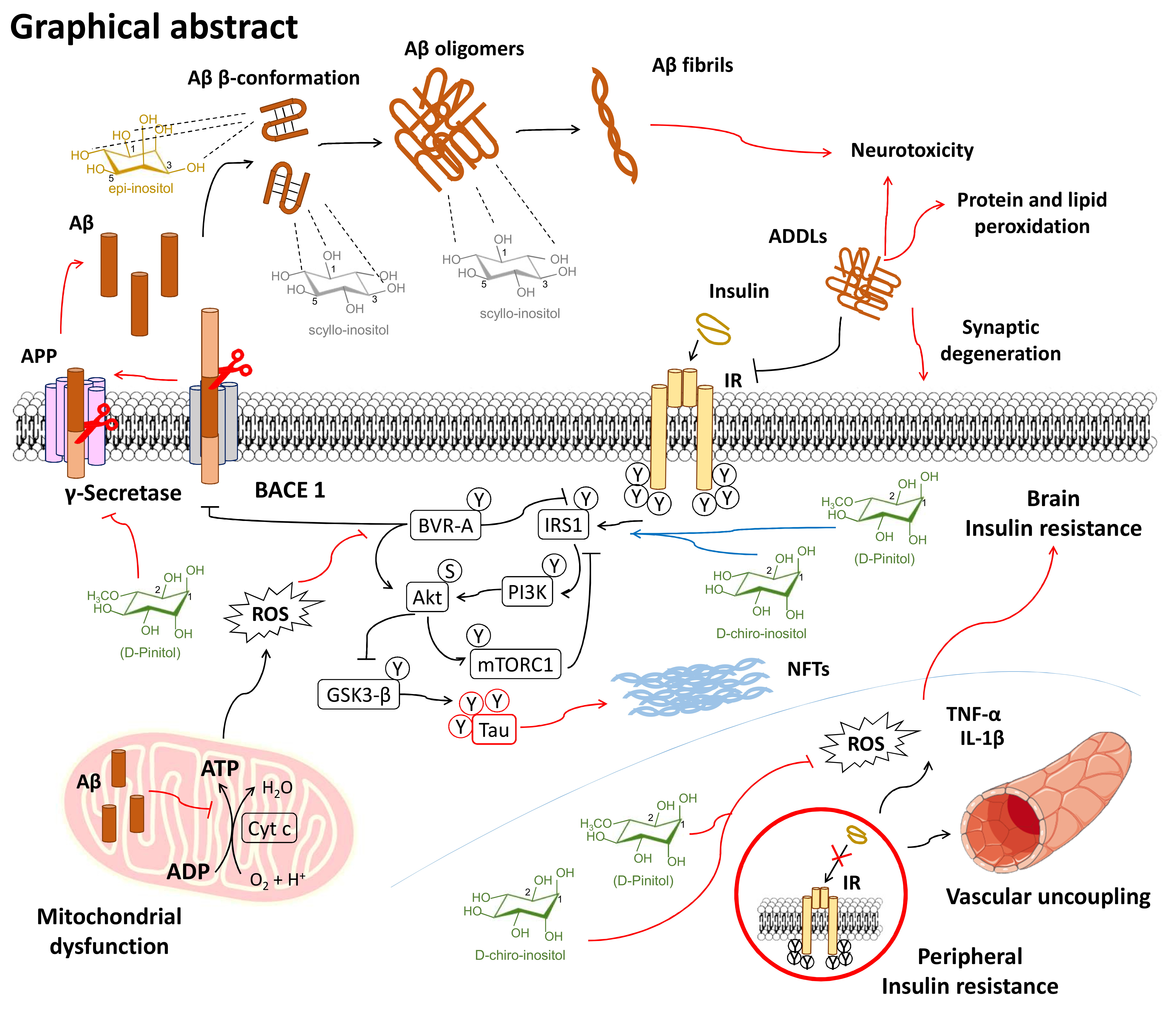{kind=link}
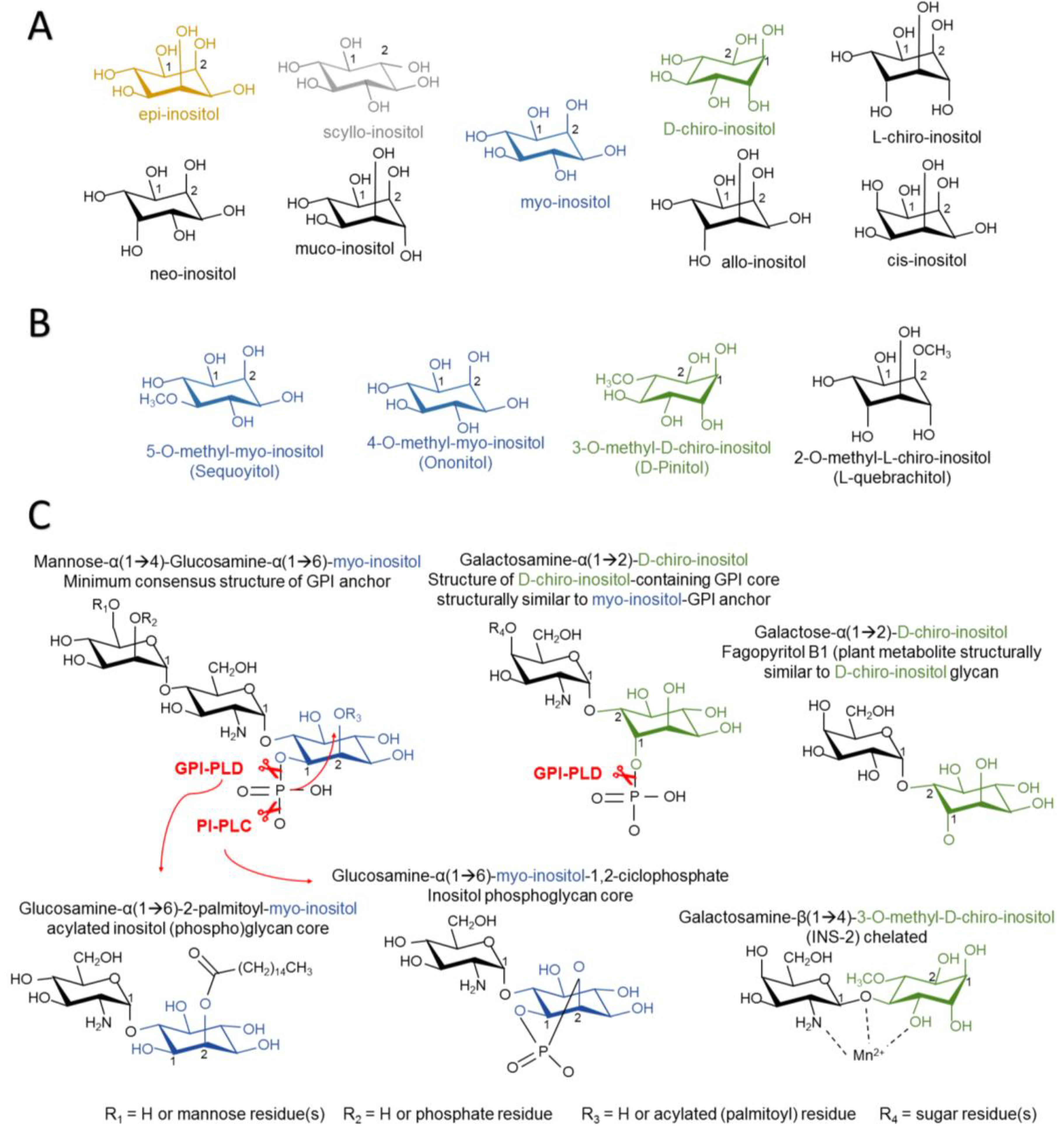{kind=link}
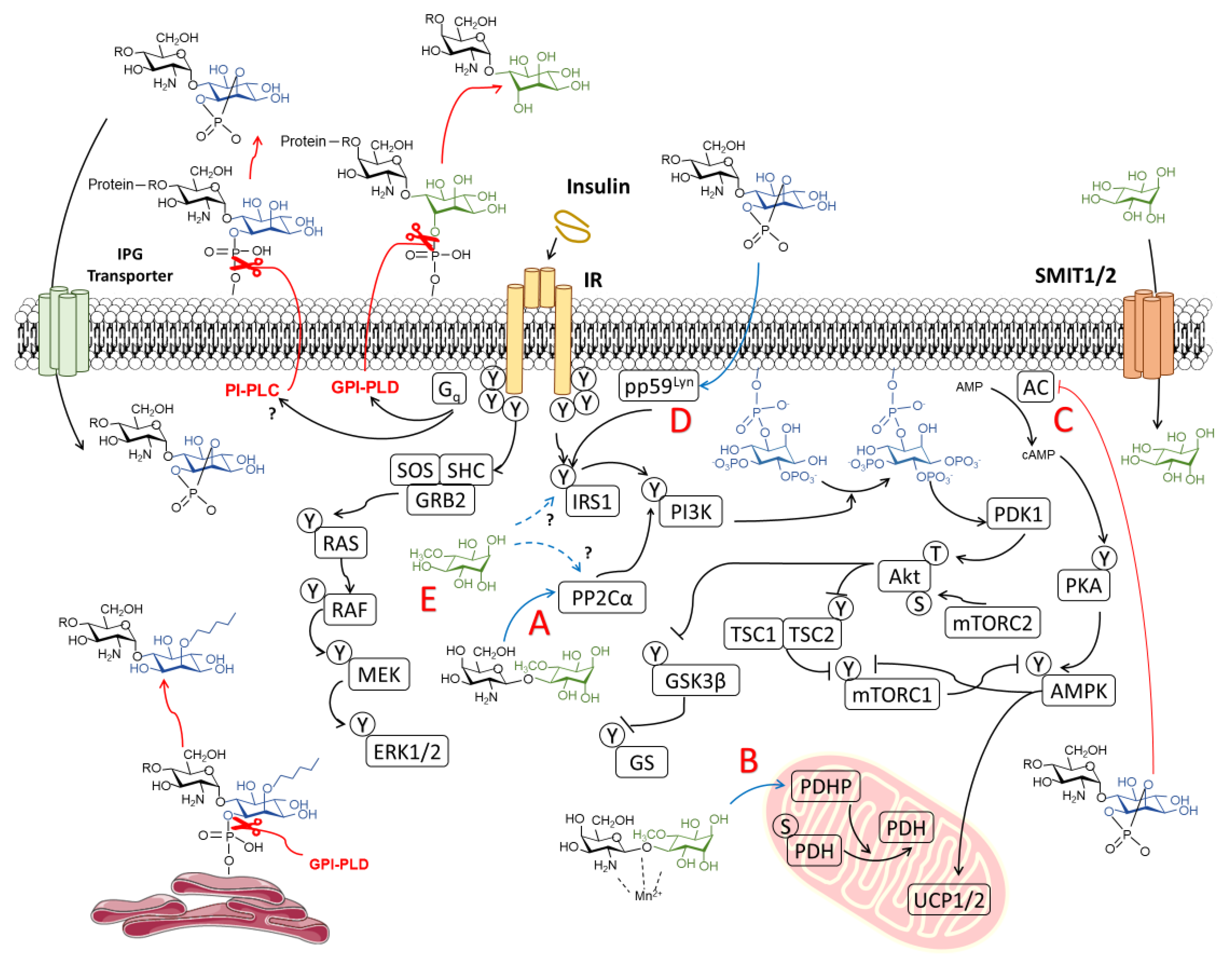{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inositol Stereoisomer | Mainly Found in | Distribution and Function in Mammals | Pharmacological Properties |
|---|---|---|---|
| Myo-inositol |
|
|
|
| Epi-inositol |
|
| |
| Scyllo-inositol |
|
|
|
| Neo-inositol |
|
|
|
| Muco-inositol |
|
|
|
| d-chiro-inositol |
|
|
|
| l-chiro-inositol |
|
|
|
| Allo-inositol |
|
|
|
| Cis-inositol |
|
|
|
| Inositol Sources in the Brain | Inositol Isoform | Main Brain Target | Described Mechanism |
|---|---|---|---|
| Myo-inositol | Astrocytes. |
|
| LPI | GPR55 in the hippocampus. | ||
| IP3 | Endoplasmic reticulum in neurons. |
| |
| PIP2 | Potassium channels GIRK and KCNQ2/3, mainly found in the hippocampus. |
| |
| PI(3)P | GABA neurons in the hippocampus. |
| |
| PI(4,5)P2 | Ion channels found in neurons. | ||
| PI(3,5)P2 | NMDA and GluA1 channels in neurons. | ||
| PI(3,4)P2 | Neurites. |
|
| Inositol | Title, NCT Number, and Date | Dose | Population | Outcome Measures | Published Results |
|---|---|---|---|---|---|
| Scyllo-inositol (ELND005) |
| Placebo, 250 mg/kg; 1000 mg/kg; 2000 mg/kg at 78 weeks |
|
| [201] |
| Placebo, 250 mg/kg; post-Dec 2009, 2000 mg/kg; pre-Dec 2009, 48 weeks |
|
| Confidence agreement | |
| Placebo, film coated tablets, twice a day for 12 weeks |
|
| Confidence agreement | |
| Continue at same dose as previous clinical trial for 36 weeks |
|
| Confidence agreement | |
| Placebo, 250 mg/kg once a day; 250 mg/kg twice a day, 4 weeks |
|
| [234] | |
| d-pinitol (NIC5-15) |
| Placebo; dose not specified, 7 weeks |
|
| [242] |
| Placebo; dose not specified, 24 weeks |
|
| N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Gambero, A.J.; Sanjuan, C.; Serrano-Castro, P.J.; Suárez, J.; Rodríguez de Fonseca, F. The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases. Biomedicines 2020, 8, 295. https://doi.org/10.3390/biomedicines8090295
López-Gambero AJ, Sanjuan C, Serrano-Castro PJ, Suárez J, Rodríguez de Fonseca F. The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases. Biomedicines. 2020; 8(9):295. https://doi.org/10.3390/biomedicines8090295
Chicago/Turabian StyleLópez-Gambero, Antonio J., Carlos Sanjuan, Pedro Jesús Serrano-Castro, Juan Suárez, and Fernando Rodríguez de Fonseca. 2020. "The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases" Biomedicines 8, no. 9: 295. https://doi.org/10.3390/biomedicines8090295
APA StyleLópez-Gambero, A. J., Sanjuan, C., Serrano-Castro, P. J., Suárez, J., & Rodríguez de Fonseca, F. (2020). The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases. Biomedicines, 8(9), 295. https://doi.org/10.3390/biomedicines8090295