
Potential Roles of Sestrin2 in Alzheimer’s Disease: Antioxidation, Autophagy Promotion, and Beyond

 , , , and
, , , and 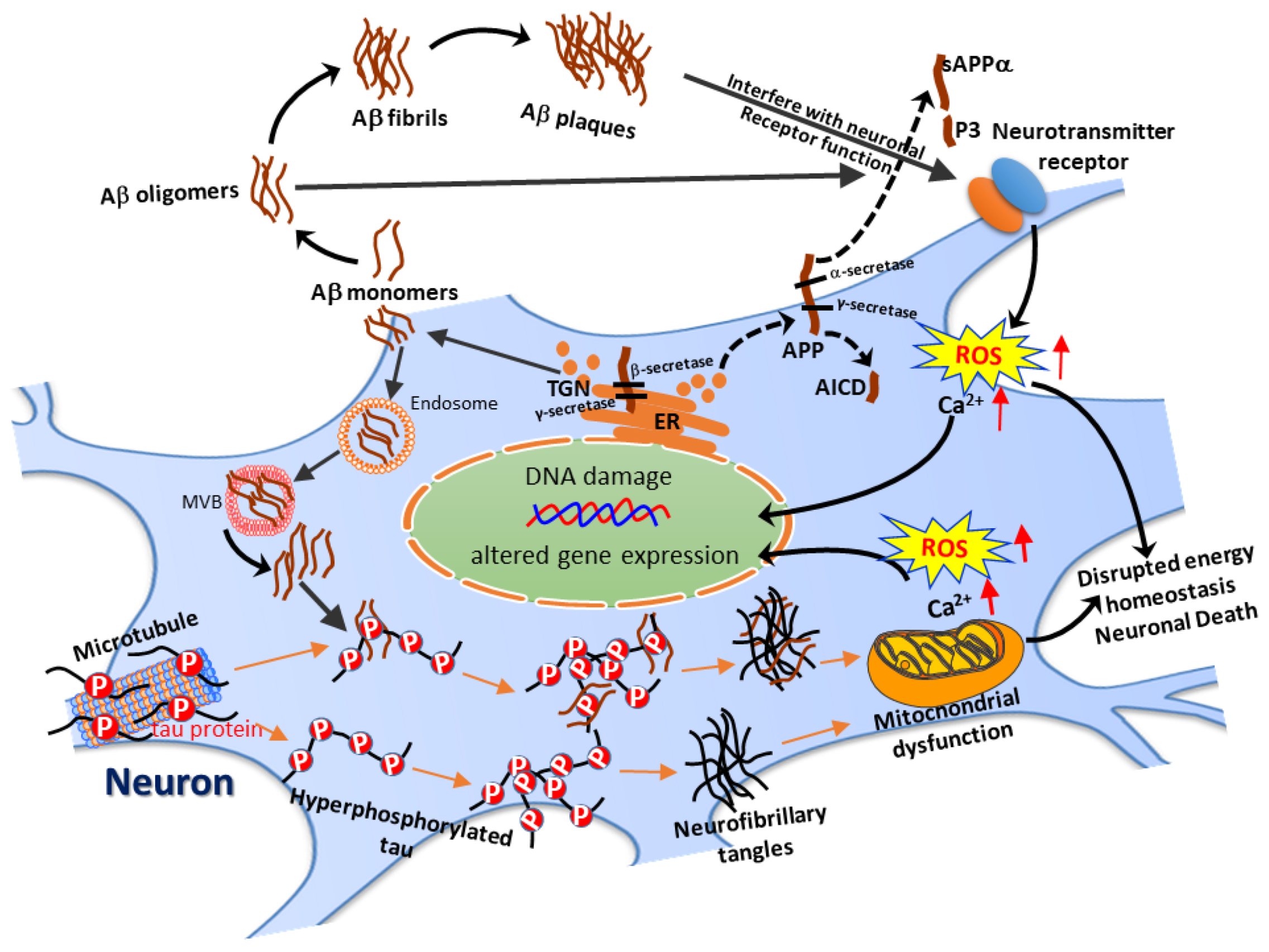{kind=link}
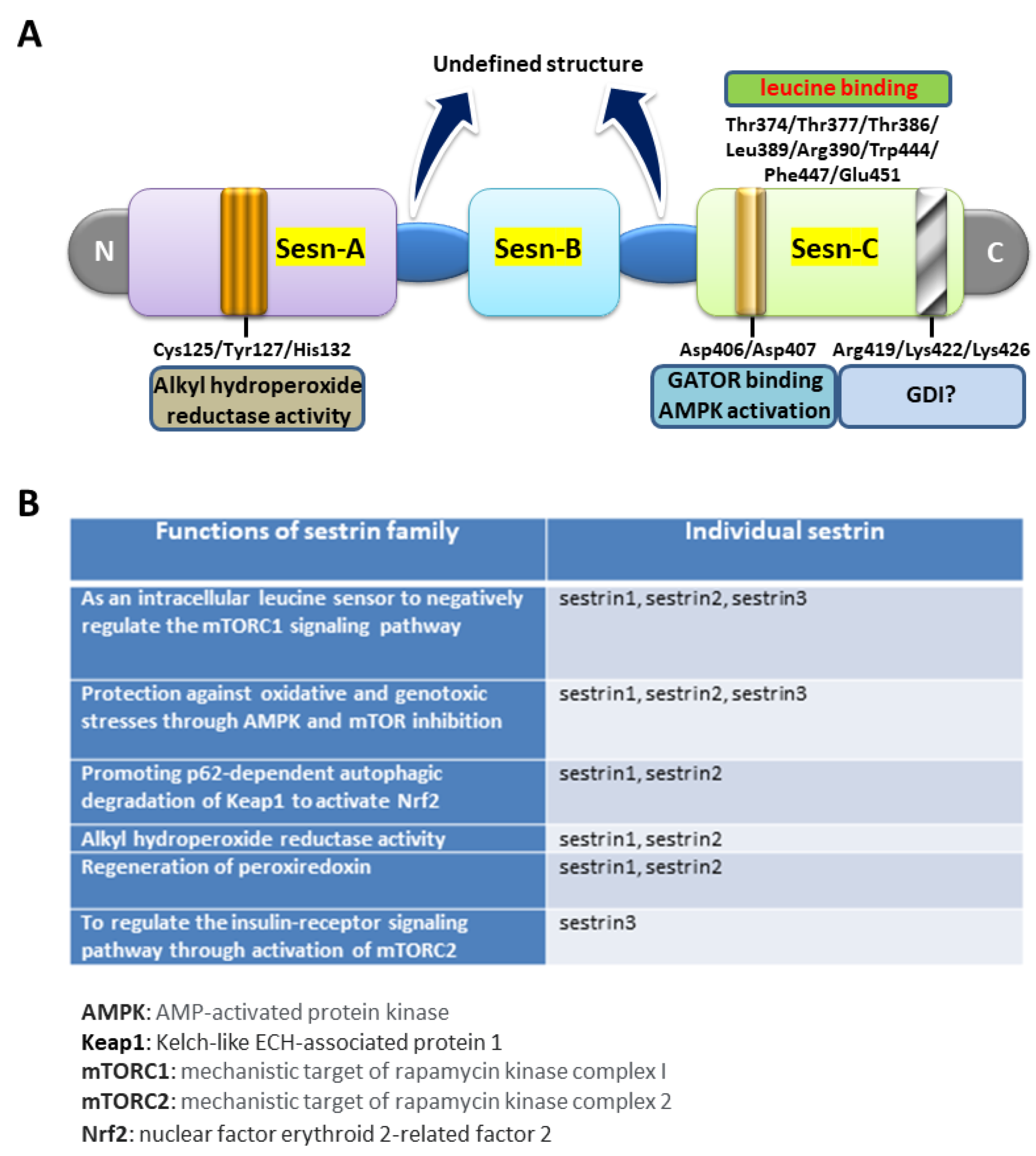{kind=link}
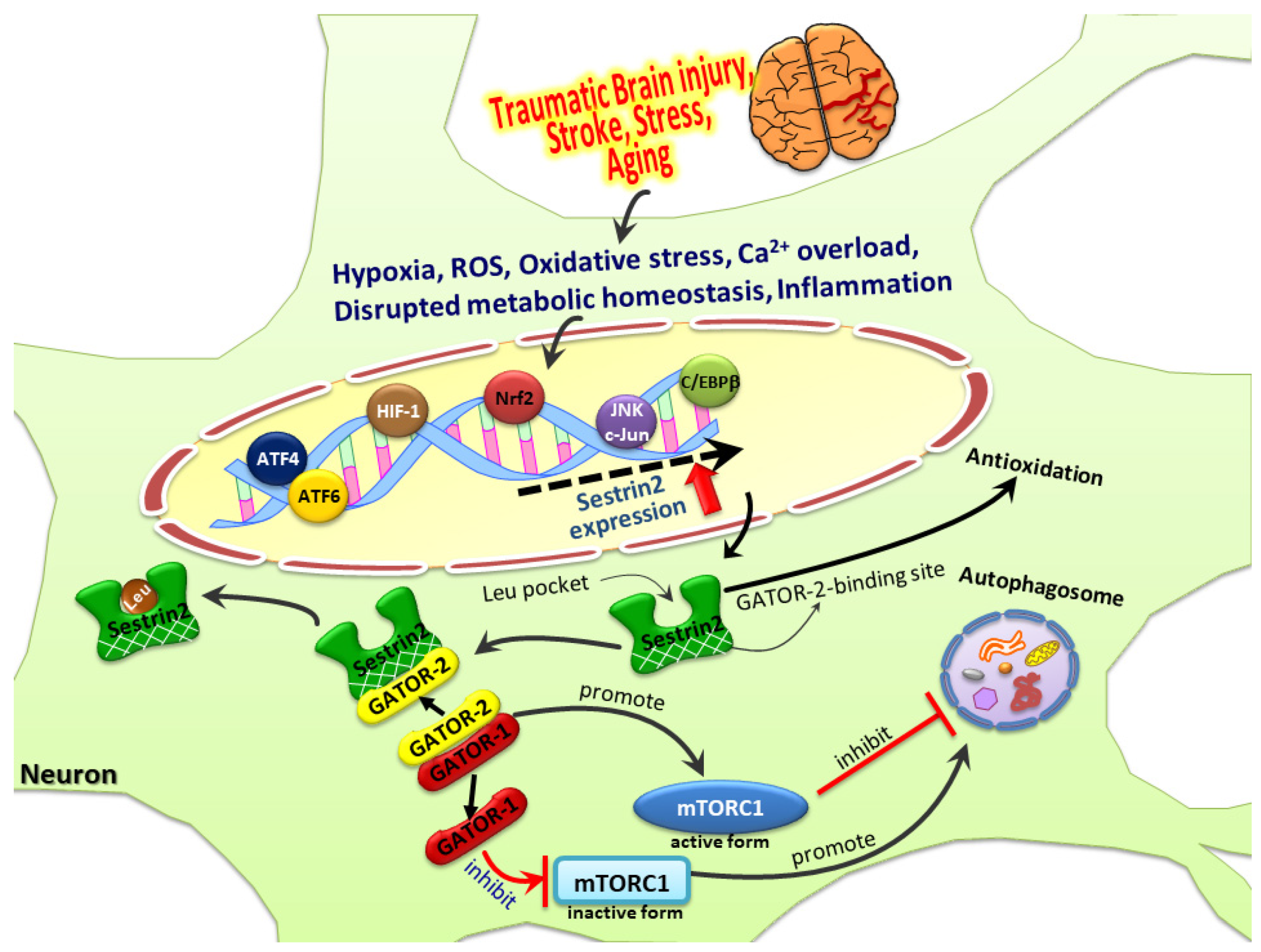{kind=link}
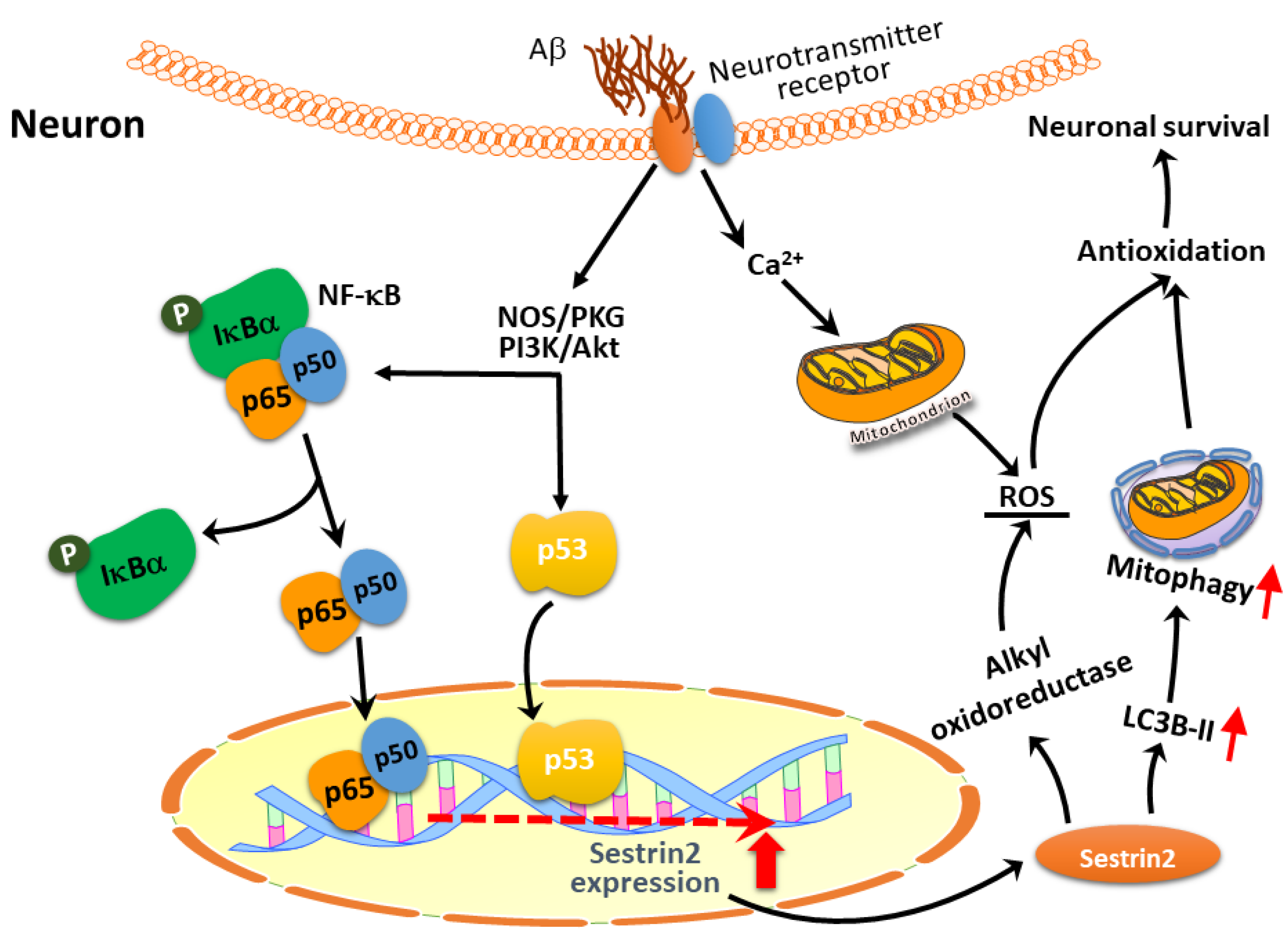{kind=link}
{kind=link}
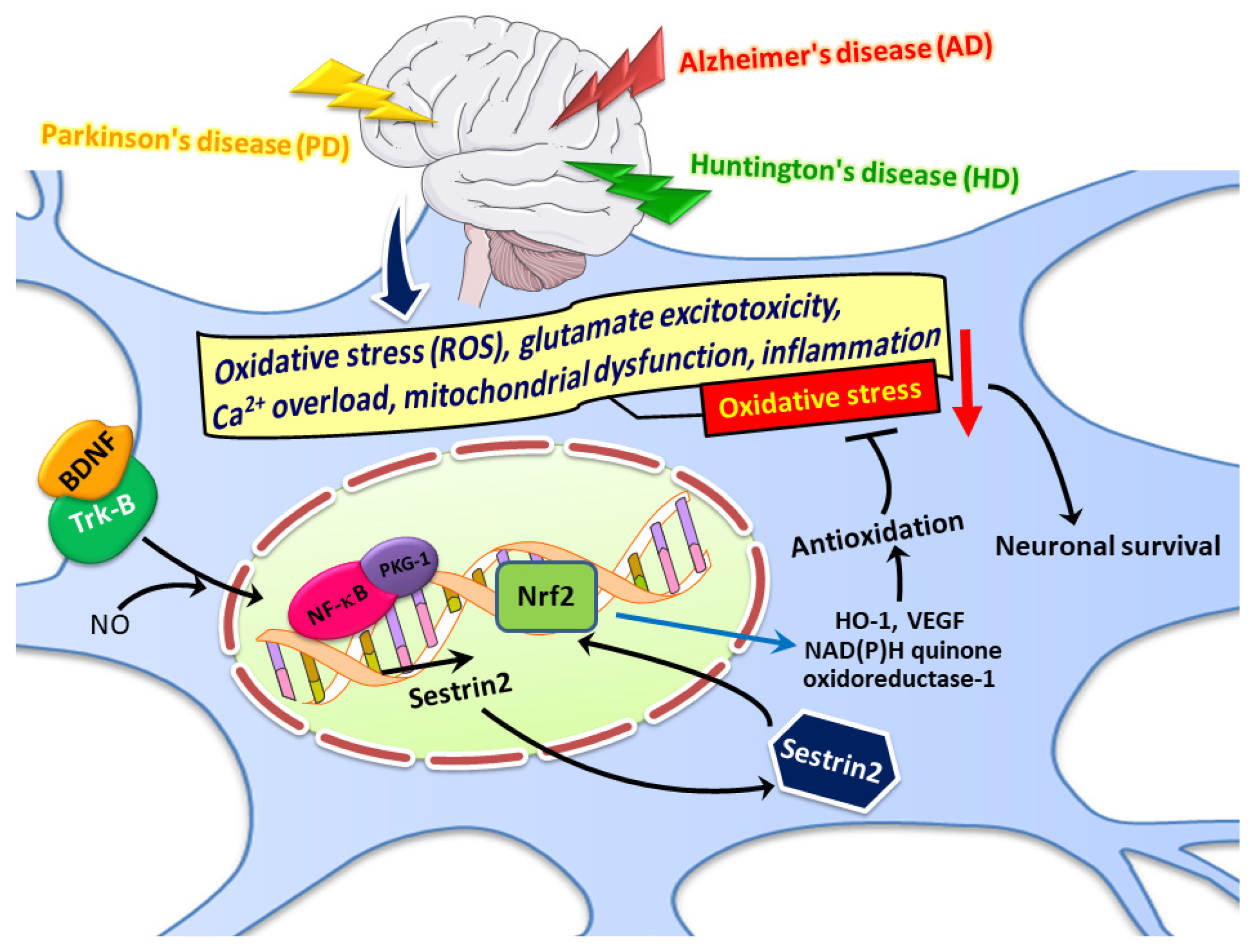
Abstract
:1. Introduction
2. The Biological Roles of Sestrin2
3. Sestrin2 in Age-Related Clinical Conditions
4. Potential Roles of Sestrin2 in Age-Related Neurodegenerative Diseases: Focusing on AD
5. Medications or Chemical Compounds Capable of Altering Sestrin2 Expression
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Takizawa, C.; Thompson, P.L.; van Walsem, A.; Faure, C.; Maier, W.C. Epidemiological and Economic Burden of Alzheimer’s Disease: A Systematic Literature Review of Data across Europe and the United States of America. J. Alzheimer’s Dis. 2014, 43, 1271–1284. [Google Scholar] [CrossRef]
- Hung, C.-W.; Chen, Y.-C.; Hsieh, W.-L.; Chiou, S.-H.; Kao, C.-L. Ageing and neurodegenerative diseases. Ageing Res. Rev. 2010, 9, S36–S46. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef] [PubMed]
- Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 2020, 26, S177–S183. [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Octave, J.N. The amyloid peptide precursor in Alzheimer’s disease. Acta Neurol. Belg. 1995, 95, 197–209. [Google Scholar] [CrossRef]
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J.; et al. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nat. Cell Biol. 1993, 361, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nat. Cell Biol. 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Tjernberg, L.; Schedin-Weiss, S. Neuronal Trafficking of the Amyloid Precursor Protein—What Do We Really Know? BioMed 2021, 9, 801. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. Amyloid beta, glutamate, excitotoxicity in Alzheimer’s disease: Are we on the right track? CNS Neurosci. Ther. 2013, 19, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.C.; Chen, S.D.; Liu, C.C.; Yang, D.I. Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic. Biol. Med. 2005, 38, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.C.; Chen, C.H.; Wu, M.H.; Hou, B.Y.; Yang, D.I. Roles of Id1/HIF-1 and CDK5/HIF-1 in cell cycle reentry induced by amyloid-beta peptide in post-mitotic cortical neuron. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118628. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.-C.; Chen, C.-H.; Chang, S.-H.; Huang, C.-T.; Hwang, W.-C.; Yang, D.-I. Id1 and Sonic Hedgehog Mediate Cell Cycle Reentry and Apoptosis Induced by Amyloid Beta-Peptide in Post-mitotic Cortical Neurons. Mol. Neurobiol. 2018, 56, 465–489. [Google Scholar] [CrossRef]
- Caldeira, G.L.; Ferreira, I.L.; Rego, A.C. Impaired Transcription in Alzheimer’s Disease: Key Role in Mitochondrial Dysfunction and Oxidative Stress. J. Alzheimer’s Dis. 2013, 34, 115–131. [Google Scholar] [CrossRef]
- Wu, M.-F.; Yin, J.-H.; Hwang, C.-S.; Tang, C.-M.; Yang, D.-I. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef]
- Vicario-Orri, E.; Opazo, C.M.; Muñoz, F.J. The Pathophysiology of Axonal Transport in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1097–1113. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Q.; Sawa, M.; Mobley, W.C. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic. Biol. Med. 2018, 114, 52–61. [Google Scholar] [CrossRef]
- Lauretti, E.; Praticò, D. Alzheimer’s disease: Phenotypic approaches using disease models and the targeting of tau protein. Expert Opin. Ther. Targets 2020, 24, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Niewiadomski, W.; Steczkowska, M.; Gasiorowska, A. Tau Oligomers Neurotoxicity. Life 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Perić, A.; Annaert, W. Early etiology of Alzheimer’s disease: Tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef] [Green Version]
- Brewer, G.J.; Herrera, R.A.; Philipp, S.; Sosna, J.; Reyes-Ruiz, J.M.; Glabe, C.G. Age-related intraneuronal aggregation of amyloid-beta in endosomes, mitochondria, autophagosomes, and lysosomes. J. Alzheimers Dis. 2020, 73, 229–246. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. B 2019, 7, 7267–7282. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Yuan, R.Y.; Hu, C.J.; Hsu, C.Y. Amyloid-beta peptide alteration of tau exon-10 splicing via the GSK3beta-SC35 pathway. Neurobiol. Dis. 2010, 40, 378–385. [Google Scholar] [CrossRef]
- Sayas, C.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P. Regeneration of Peroxiredoxins by p53-Regulated Sestrins, Homologs of Bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, H.; Debeer, P.; Bairoch, A.; Wilquet, V.; Huysmans, C.; Parthoens, E.; Fryns, J.P.; Gewillig, M.; Nakamura, Y.; Niikawa, N.; et al. PA26 is a candidate gene for heterotaxia in humans: Identification of a novel PA26-related gene family in human and mouse. Qual. Life Res. 2003, 112, 573–580. [Google Scholar] [CrossRef]
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-Miguel, S.; Buckbinder, L.; Jean, P.; Gelbert, L.; Talbott, R.; Laidlaw, J.; Seizinger, B.; Kley, N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999, 18, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; An, S.; Ro, S.-H.; Teixeira, F.; Park, G.J.; Kim, C.; Cho, C.-S.; Kim, J.-S.; Jakob, U.; Lee, J.H.; et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat. Commun. 2015, 6, 10025. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2015, 351, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Haidurov, A.; Budanov, A.V. Sestrin family—The stem controlling healthy ageing. Mech. Ageing Dev. 2020, 192, 111379. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins Orchestrate Cellular Metabolism to Attenuate Aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-S.; Chen, S.-D.; Wu, C.-L.; Huang, S.-S.; Yang, D.-I. Induction of sestrin2 as an endogenous protective mechanism against amyloid beta-peptide neurotoxicity in primary cortical culture. Exp. Neurol. 2014, 253, 63–71. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Chao, A.C.; Lin, Y.C.; Chen, S.D.; Yang, D.I. The p53/NF-kappaB-dependent induction of sestrin2 by amyloid-beta peptides exerts antioxidative actions in neurons. Free Radic. Biol. Med. 2021, 169, 36–61. [Google Scholar] [CrossRef]
- Buckbinder, L.; Talbott, R.; Seizinger, B.R.; Kley, N. Gene regulation by temperature-sensitive p53 mutants: Identification of p53 response genes. Proc. Natl. Acad. Sci. USA 1994, 91, 10640–10644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Sablina, A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; E Kravchenko, J.; Chumakov, P. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, A.; Budanov, A. Sensing the Environment Through Sestrins: Implications for Cellular Metabolism. Pancreat. ß-Cell Biol. Health Dis. 2016, 327, 1–42. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2015, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting p62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Xi, Y.; Chen, K.; Xiao, P.; Li, S.; Sun, X.; Han, Z. Upregulation Sestrin2 protects against hydrogen peroxide-induced oxidative damage bovine mammary epithelial cells via a Keap1-Nrf2/ARE pathway. J. Cell. Physiol. 2021, 236, 392–404. [Google Scholar] [CrossRef]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef]
- Olson, N.; Hristova, M.; Heintz, N.H.; Lounsbury, K.M.; Van Der Vliet, A. Activation of hypoxia-inducible factor-1 protects airway epithelium against oxidant-induced barrier dysfunction. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L993–L1002. [Google Scholar] [CrossRef] [Green Version]
- Essler, S.; Dehne, N.; Brüne, B. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009, 583, 3531–3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a Novel Hypoxia-Inducible Factor 1-Responsive Gene, RTP801, Involved in Apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Nuclear factor-kappaB-dependent Sestrin2 induction mediates the antioxidant effects of BDNF against mitochondrial inhibition in rat cortical neurons. Mol. Neurobiol. 2016, 53, 4126–4142. [Google Scholar] [CrossRef]
- Liu, J.; Amar, F.; Corona, C.; So, R.; Andrews, S.J.; Nagy, P.L.; Shelanski, M.L.; Greene, L.A. Brain-Derived Neurotrophic Factor Elevates Activating Transcription Factor 4 (ATF4) in Neurons and Promotes ATF4-Dependent Induction of Sesn2. Front. Mol. Neurosci. 2018, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaeva, A.; Kovaleva, I.E.; Chumakov, P.M.; Evstafieva, A.G. Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4. Cell Cycle 2016, 15, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chen, X.A.; Hu, J.; Jiang, J.-K.; Li, Y.; Chan-Salis, K.Y.; Gu, Y.; Chen, G.; Thomas, C.; Pugh, B.F.; et al. ATF4 Gene Network Mediates Cellular Response to the Anticancer PAD Inhibitor YW3-56 in Triple-Negative Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Palm, W.; Peng, M.; King, B.; Lindsten, T.; Li, M.; Koumenis, C.; Thompson, C.B. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015, 29, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Wu, X.-Q.; Deng, R.; Sun, T.; Feng, G.-K.; Zhu, X.-F. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell. Signal. 2013, 25, 150–158. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Jegal, K.H.; Park, S.M.; Cho, S.S.; Byun, S.H.; Ku, S.K.; Kim, S.C.; Ki, S.H.; Cho, I.J. Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury. Biochim. Biophys. Acta (BBA) Bioenerget. 2017, 1864, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-W.; Park, H.; Ro, S.-H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Bodmer, R.; Bier, E.; Karin, M. Sestrins at the crossroad between stress and aging. Aging 2010, 2, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Budanov, A.V.; Lee, J.H.; Karin, M. Stressin’ Sestrins take an aging fight. EMBO Mol. Med. 2010, 2, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-B.; Xuan, Y.; Shi, W.-J.; Chi, F.; Xing, R.; Zeng, Y.-C. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer. Am. J. Transl. Res. 2016, 8, 1903–1909. [Google Scholar]
- Wei, J.-L.; Fu, Z.-X.; Fang, M.; Guo, J.-B.; Zhao, Q.-N.; Lu, W.-D.; Zhou, Q.-Y. Decreased expression of sestrin 2 predicts unfavorable outcome in colorectal cancer. Oncol. Rep. 2014, 33, 1349–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Kim, D.Y.; Kang, S.H.; Yun, H.K.; Kim, J.L.; Kim, B.R.; Park, S.H.; Na, Y.J.; Jo, M.J.; Jeong, Y.A.; et al. Docosahexaenoic Acid Enhances Oxaliplatin-Induced Autophagic Cell Death via the ER Stress/Sesn2 Pathway in Colorectal Cancer. Cancers 2019, 11, 982. [Google Scholar] [CrossRef] [Green Version]
- Ro, S.-H.; Xue, X.; Ramakrishnan, S.K.; Cho, C.-S.; Namkoong, S.; Jang, I.; A Semple, I.; Ho, A.; Park, H.-W.; Shah, Y.M.; et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. eLife 2016, 5, e12204. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.-W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of Metabolic Homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundararajan, S.; Jayachandran, I.; Subramanian, S.C.; Anjana, R.M.; Balasubramanyam, M.; Mohan, V.; Venkatesan, B.; Manickam, N. Decreased Sestrin levels in patients with type 2 diabetes and dyslipidemia and their association with the severity of atherogenic index. J. Endocrinol. Investig. 2021, 44, 1395–1405. [Google Scholar] [CrossRef]
- Mohany, K.M.; Al Rugaie, O. Association of serum sestrin 2 and betatrophin with serum neutrophil gelatinase associated lipocalin levels in type 2 diabetic patients with diabetic nephropathy. J. Diabetes Metab. Disord. 2020, 19, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-H.; Ruan, J.-Y.; Liu, H.-J.; Liu, Y.; Feng, H.; Tang, Q.-Z. Sestrin family may play important roles in the regulation of cardiac pathophysiology. Int. J. Cardiol. 2016, 202, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Du, X.; Huang, Z.; Quan, N. Sestrin 2, a potential star of antioxidant stress in cardiovascular diseases. Free. Radic. Biol. Med. 2021, 163, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, N.; Shao, X.; Li, J.; Guo, L.; Yu, X.; Sun, Y.; Hao, J.; Niu, H.; Xiang, J.; et al. Increased plasma sestrin2 concentrations in patients with chronic heart failure and predicted the occurrence of major adverse cardiac events: A 36-month follow-up cohort study. Clin. Chim. Acta 2019, 495, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Li, W.; Song, Y.; Li, Z.; Tang, Y.; Zhang, Z.; Yang, G.Y. Sestrin2 regulates microglia polarization through mTOR-mediated autophagic flux to attenuate inflammation during experimental brain ischemia. J. Neuroinflamm. 2020, 17, 329. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Zhou, Y.; Wang, P.; Li, L.; Zhao, Y. Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res. 2020, 1745, 146948. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, Y.; Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Li, L.; Zhao, Y. Sestrin2 overexpression attenuates focal cerebral ischemic injury in rat by increasing Nrf2/HO-1 pathway-mediated angiogenesis. Neuroscience 2019, 410, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiao, L.; Hou, Y.; He, Q.; Zhu, J.; Li, Y.; Wu, J.; Zhao, J.; Yu, S.; Zhao, Y. Sestrin2 Silencing Exacerbates Cerebral Ischemia/Reperfusion Injury by Decreasing Mitochondrial Biogenesis through the AMPK/PGC-1α Pathway in Rats. Sci. Rep. 2016, 6, 30272. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Chen, R. Sestrin1 exerts a cytoprotective role against oxygen-glucose deprivation/reoxygenation-induced neuronal injury by potentiating Nrf2 activation via the modulation of Keap1. Brain Res. 2021, 1750, 147165. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.R.; Behmoaras, J.; Bottolo, L.; Krishnan, M.L.; Pernhorst, K.; Santoscoy, P.L.M.; Rossetti, T.; Speed, D.; Srivastava, P.K.; Chadeau-Hyam, M.; et al. Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisari, F.; Roncon, P.; Soukoupova, M.; Paolone, G.; Labasque, M.; Ingusci, S.; Falcicchia, C.; Marino, P.; Johnson, M.; Rossetti, T.; et al. Implication of sestrin3 in epilepsy and its comorbidities. Brain Commun. 2021, 3, fcaa130. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.G.; Zou, J.; Lu, Q.C. Silencing rno-miR-155-5p in rat temporal lobe epilepsy model reduces pathophysiological features and cell apoptosis by activating Sestrin-3. Brain Res. 2018, 1689, 109–122. [Google Scholar] [CrossRef]
- Numakawa, T.; Matsumoto, T.; Numakawa, Y.; Richards, M.; Yamawaki, S.; Kunugi, H. Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration. J. Toxicol. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Rocha, E.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution ofα-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhan, C.; Zhong, Q.; Li, S. Upregulation of sestrin-2 expression via P53 protects against 1-methyl-4-phenylpyridinium (MPP+) neurotoxicity. J. Mol. Neurosci. 2013, 51, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-S.; Guan, J.-J.; Xu, H.-D.; Wu, F.; Sheng, R.; Qin, Z.-H. Sestrin2 Protects Dopaminergic Cells against Rotenone Toxicity through AMPK-Dependent Autophagy Activation. Mol. Cell. Biol. 2015, 35, 2740–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, N.; Upadhyay, A.D.; Goyal, V.; Dwivedi, S.; Dey, A.B.; Dey, S. Sestrin2 as Serum Protein Marker and Potential Therapeutic Target for Parkinson’s Disease. J. Gerontol. Ser. A: Boil. Sci. Med. Sci. 2019, 75, 690–695. [Google Scholar] [CrossRef]
- Chen, S.-D.; Wu, C.-L.; Hwang, W.-C.; Yang, D.-I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunez, I.; Tasset, I.; Perez-De La Cruz, V.; Santamaria, A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: Past, present and future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-L.; Hwang, C.-S.; Chen, S.-D.; Yin, J.; Yang, D.-I. Neuroprotective mechanisms of brain-derived neurotrophic factor against 3-nitropropionic acid toxicity: Therapeutic implications for Huntington’s disease. Ann. N. Y. Acad. Sci. 2010, 1201, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-D.; Yang, J.-L.; Lin, T.-K.; Yang, D.-I. Emerging Roles of Sestrins in Neurodegenerative Diseases: Counteracting Oxidative Stress and Beyond. J. Clin. Med. 2019, 8, 1001. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Karahan, H.; Kelicen-Ugur, P. Effect of atorvastatin on Abeta1-42-induced alteration of SESN2, SIRT1, LC3II and TPP1 protein expressions in neuronal cell cultures. J. Pharm. Pharmacol. 2020, 72, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.; Kumar, R.; Desai, G.R.; Venugopalan, G.; Shekhar, S.; Chatterjee, P.; Tripathi, M.; Upadhyay, A.D.; Dwivedi, S.; Dey, A.B.; et al. Relative Alterations in Blood-Based Levels of Sestrin in Alzheimer’s Disease and Mild Cognitive Impairment Patients. J. Alzheimer’s Dis. 2016, 54, 1147–1155. [Google Scholar] [CrossRef]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soontornniyomkij, V.; Soontornniyomkij, B.; Moore, D.J.; Gouaux, B.; Masliah, E.; Tung, S.; Vinters, H.V.; Grant, I.; Achim, C.L. Antioxidant Sestrin-2 Redistribution to Neuronal Soma in Human Immunodeficiency Virus-Associated Neurocognitive Disorders. J. Neuroimmune Pharmacol. 2012, 7, 579–590. [Google Scholar] [CrossRef]
- Kim, J.R.; Lee, S.R.; Chung, H.J.; Kim, S.; Baek, S.H.; Kim, J.H.; Kim, Y.S. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: Involvement of RTP801 in amyloid beta-peptide toxicity. Exp. Mol. Med. 2003, 35, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Tampellini, D. Synaptic activity and Alzheimer’s disease: A critical update. Front. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadia, S.; Soriano, F.; Léveillé, F.; Martel, M.-A.; A Dakin, K.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Shen, J. Function and dysfunction of presenilin. Neurodegener. Dis. 2013, 13, 61–63. [Google Scholar] [CrossRef] [Green Version]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, B.; Beglopoulos, V.; Wines-Samuelson, M.; Zhang, D.; Dragatsis, I.; Südhof, T.C.; Shen, J. Presenilins are essential for regulating neurotransmitter release. Nat. Cell Biol. 2009, 460, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.L.; Sui, H.J.; Liang, B.; Wang, H.M.; Qu, W.H.; Yu, S.X.; Jin, Y. Atorvastatin prevents amyloid-beta peptide oligomer-induced synaptotoxicity and memory dysfunction in rats through a p38 MAPK-dependent pathway. Acta Pharmacol. Sin. 2014, 35, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yang, Y.-J.; Wang, H.; Dong, Q.-T.; Wang, T.-J.; Qian, H.-Y.; Xu, H. Autophagy Activation: A Novel Mechanism of Atorvastatin to Protect Mesenchymal Stem Cells from Hypoxia and Serum Deprivation via AMP-Activated Protein Kinase/Mammalian Target of Rapamycin Pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arancibia, S.; Silhol, M.; Mouliere, F.; Meffre, J.; Höllinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.; Coppola, G.; Tsukada, S.; E Schroeder, B.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Ma, X.; Ma, L.; Li, S.; Zheng, J.; Lv, J.; Cui, L.; Lv, J. Inhibition of microRNA-148b-3p alleviates oxygen-glucose deprivation/reoxygenation-induced apoptosis and oxidative stress in HT22 hippocampal neuron via reinforcing Sestrin2/Nrf2 signalling. Clin. Exp. Pharmacol. Physiol. 2020, 47, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xing, Y.; Xiong, L.; Wang, J. Sestrin2 overexpression alleviates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells by enhancing Nrf2 activation via Keap1 downregulation. Chem. Interact. 2020, 324, 109086. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef]
- Nabirotchkin, S.; E Peluffo, A.; Rinaudo, P.; Yu, J.; Hajj, R.; Cohen, D. Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. 2020, 51, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Yang, J.-L.; Yang, Y.-R.; Chen, S.-D. The potential of drug repurposing combined with reperfusion therapy in cerebral ischemic stroke: A supplementary strategy to endovascular thrombectomy. Life Sci. 2019, 236, 116889. [Google Scholar] [CrossRef]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef]
- Ihara, M.; Saito, S. Drug Repositioning for Alzheimer’s Disease: Finding Hidden Clues in Old Drugs. J. Alzheimer’s Dis. 2020, 74, 1013–1028. [Google Scholar] [CrossRef]
- Singh, R.K. Recent Trends in the Management of Alzheimer’s Disease: Current Therapeutic Options and Drug Repurposing Approaches. Curr. Neuropharmacol. 2020, 18, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, P.; Chai, C.; Sperlágh, B.; Huleatt, P.B.; Mátyus, P. Clinical utility of neuroprotective agents in neurodegenerative diseases: Current status of drug development for Alzheimer’s, Parkinson’s and Huntington’s diseases, and amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2012, 21, 1267–1308. [Google Scholar] [CrossRef]
- Sun, X.; Han, F.; Lu, Q.; Li, X.; Ren, D.; Zhang, J.; Han, Y.; Xiang, Y.K. Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating Sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes 2020, 69, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ding, C.; Zhang, G.; Pan, R.; Liu, Y.; Huang, N.; Hou, N.; Han, F.; Xu, W.; Sun, X. Liraglutide ameliorates obesity-related nonalcoholic fatty liver disease by regulating Sestrin2-mediated Nrf2/HO-1 pathway. Biochem. Biophys. Res. Commun. 2020, 525, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Entezar-Almahdi, E.; Mohammadi-Samani, S.; Tayebi, L.; Farjadian, F. Recent Advances in Designing 5-Fluorouracil Delivery Systems: A Stepping Stone in the Safe Treatment of Colorectal Cancer. Int. J. Nanomed. 2020, 15, 5445–5458. [Google Scholar] [CrossRef]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Seo, K.; Ki, S.H.; Park, E.Y.; Shin, S.M. 5-Fluorouracil inhibits cell migration by induction of Sestrin2 in colon cancer cells. Arch. Pharmacal Res. 2016, 40, 231–239. [Google Scholar] [CrossRef]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef]
- Lan, X.; Kiyota, T.; Hanamsagar, R.; Huang, Y.; Andrews, S.; Peng, H.; Zheng, J.C.; Swindells, S. The effect of HIV protease inhibitors on amyloid-beta peptide degradation and synthesis in human cells and Alzheimer’s disease animal model. J. Neuroimmune Pharmacol. 2012, 7, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Byun, J.; Park, M.; Kim, S.W.; Lee, S.; Kim, J.; Lee, I.; Choi, Y.; Park, K. Melatonin inhibits vascular smooth muscle cell proliferation and apoptosis through upregulation of Sestrin2. Exp. Ther. Med. 2020, 19, 3454–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.; Seo, S.; Han, J.Y.; Ki, S.H.; Shin, S.M. Resveratrol attenuates methylglyoxal-induced mitochondrial dysfunction and apoptosis by Sestrin2 induction. Toxicol. Appl. Pharmacol. 2014, 280, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; Yang, J.H.; Shin, B.Y.; Seo, K.; Shin, S.M.; Cho, I.J.; Ki, S.H. Resveratrol inhibits LXRalpha-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol. Appl. Pharmacol. 2013, 271, 95–105. [Google Scholar] [CrossRef]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly)phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects Against Chronic Diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a Therapeutic Agent for Neurodegenerative Diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Malaguarnera, L. Influence of Resveratrol on the Immune Response. Nutrients 2019, 11, 946. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.; Wu, J.; Liang, J.; Yang, C.; Wang, K.; Wang, J.; Guo, X. Eupatilin protects chondrocytes from apoptosis via activating sestrin2-dependent autophagy. Int. Immunopharmacol. 2019, 75, 105748. [Google Scholar] [CrossRef]
- Jegal, K.H.; Ko, H.L.; Park, S.M.; Byun, S.H.; Kang, K.W.; Cho, I.J.; Kim, S.C. Eupatilin induces Sestrin2-dependent autophagy to prevent oxidative stress. Apoptosis 2016, 21, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-X.; Wu, J.-Z.; Li, Z.; Zhang, C.; Shi, M.-T.; Zhao, J.; Jin, M.-W.; Liu, H. Pentamethylquercetin protects against cardiac remodeling via activation of Sestrin2. Biochem. Biophys. Res. Commun. 2019, 512, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Shin, B.Y.; Han, J.Y.; Kim, M.G.; Wi, J.E.; Kim, Y.W.; Cho, I.J.; Kim, S.C.; Shin, S.M.; Ki, S.H. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol. Appl. Pharmacol. 2014, 274, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Sharopov, F.; Fokou, P.V.T.; Kobylinska, A.; De Jonge, L.; Tadio, K.; Sharifi-Rad, J.; Posmyk, M.M.; Martorell, M.; Martins, N.; et al. Melatonin in Medicinal and Food Plants: Occurrence, Bioavailability, and Health Potential for Humans. Cells 2019, 8, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-D.; Yang, J.-L.; Hsieh, Y.-H.; Lin, T.-K.; Lin, Y.-C.; Chao, A.-C.; Yang, D.-I. Potential Roles of Sestrin2 in Alzheimer’s Disease: Antioxidation, Autophagy Promotion, and Beyond. Biomedicines 2021, 9, 1308. https://doi.org/10.3390/biomedicines9101308
Chen S-D, Yang J-L, Hsieh Y-H, Lin T-K, Lin Y-C, Chao A-C, Yang D-I. Potential Roles of Sestrin2 in Alzheimer’s Disease: Antioxidation, Autophagy Promotion, and Beyond. Biomedicines. 2021; 9(10):1308. https://doi.org/10.3390/biomedicines9101308
Chicago/Turabian StyleChen, Shang-Der, Jenq-Lin Yang, Yi-Heng Hsieh, Tsu-Kung Lin, Yi-Chun Lin, A-Ching Chao, and Ding-I Yang. 2021. "Potential Roles of Sestrin2 in Alzheimer’s Disease: Antioxidation, Autophagy Promotion, and Beyond" Biomedicines 9, no. 10: 1308. https://doi.org/10.3390/biomedicines9101308
APA StyleChen, S. -D., Yang, J. -L., Hsieh, Y. -H., Lin, T. -K., Lin, Y. -C., Chao, A. -C., & Yang, D. -I. (2021). Potential Roles of Sestrin2 in Alzheimer’s Disease: Antioxidation, Autophagy Promotion, and Beyond. Biomedicines, 9(10), 1308. https://doi.org/10.3390/biomedicines9101308