
Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases

 , , ,
, , ,  ,
, 
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Transcriptome Analysis
2.3. Metabolic Analysis
2.4. Network Analysis
2.5. Zebrafish Data Acquisition and Analysis
2.6. Data and Code Accessibility
2.7. Ethics Statement
3. Results
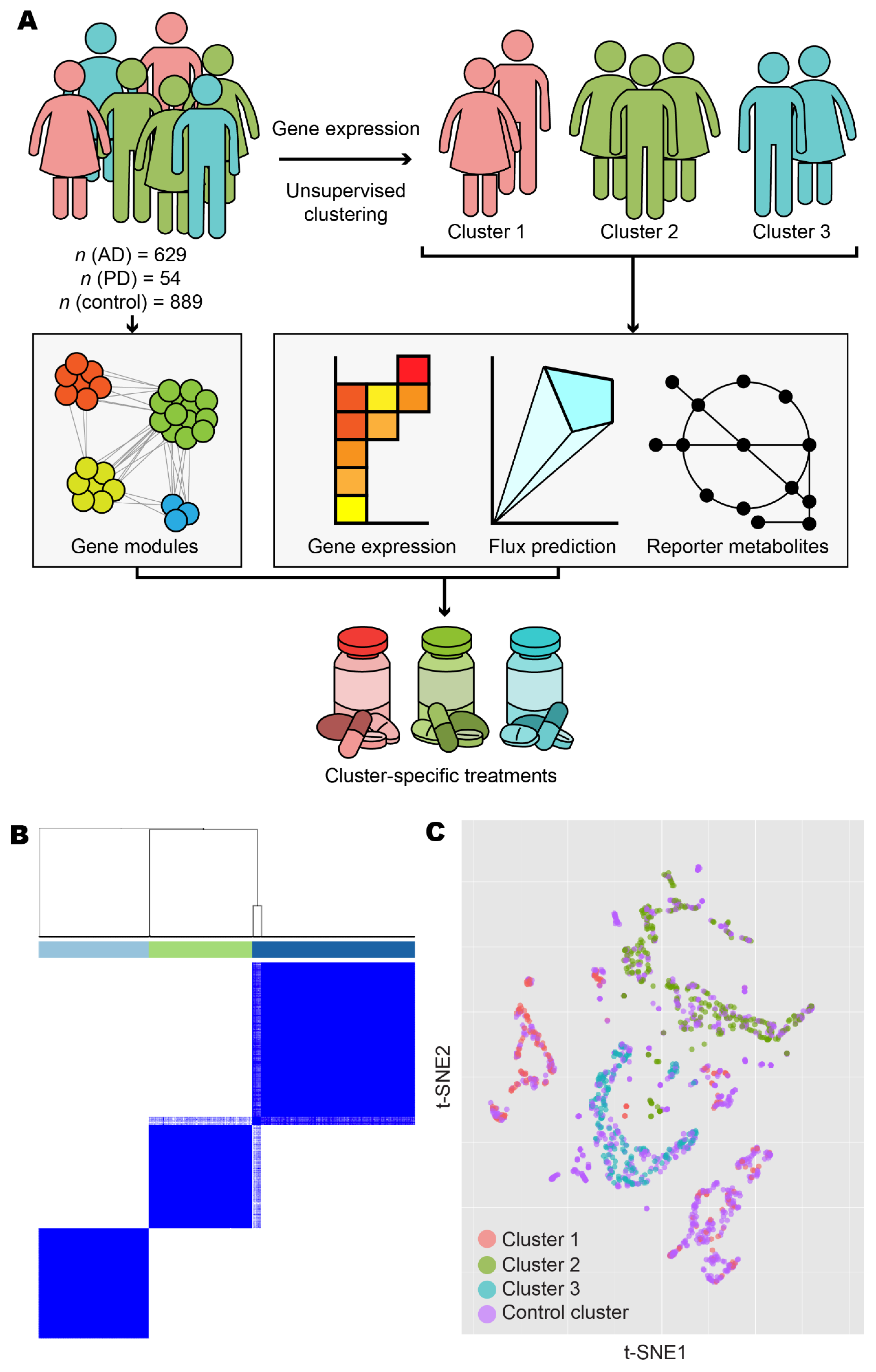
3.1. Stratification of Patients Revealed Three Distinct Disease Classes
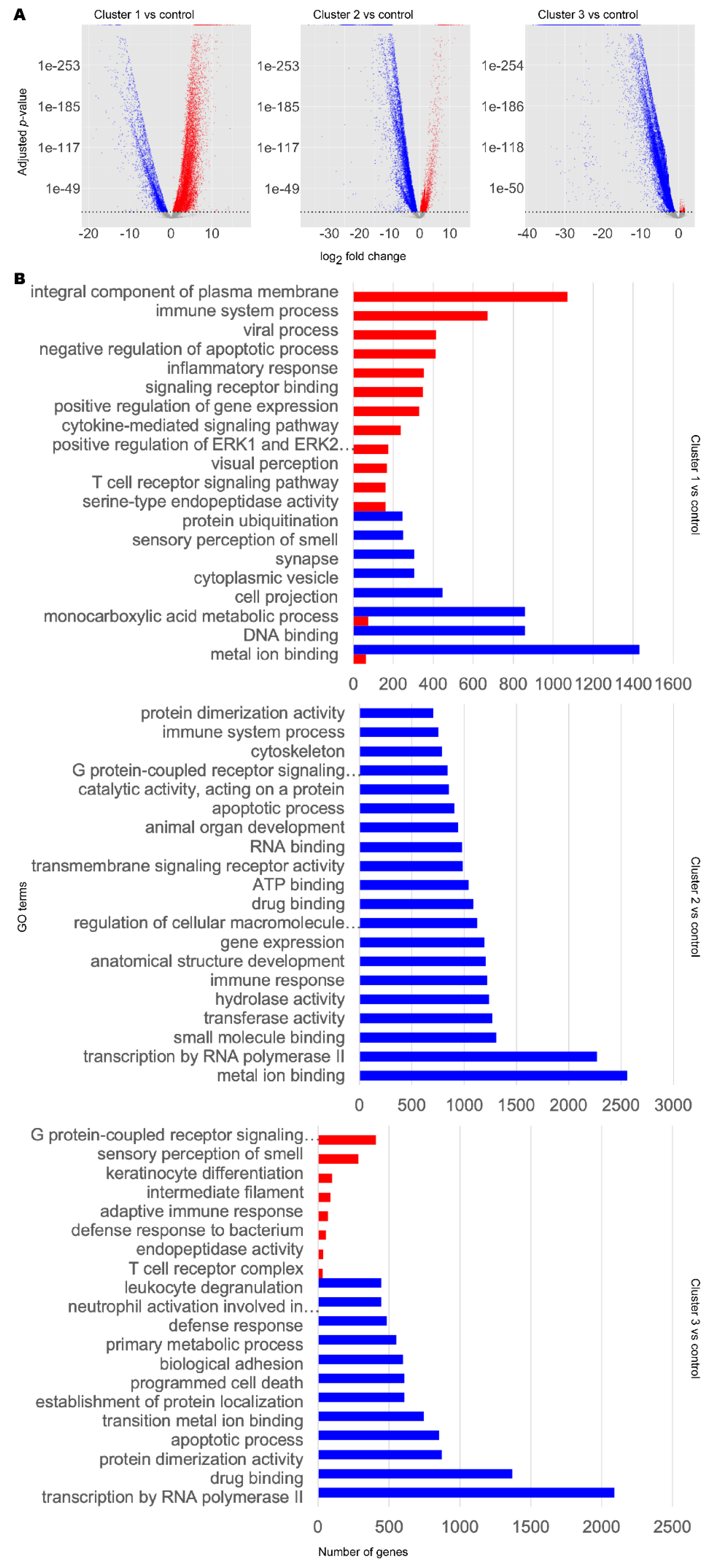
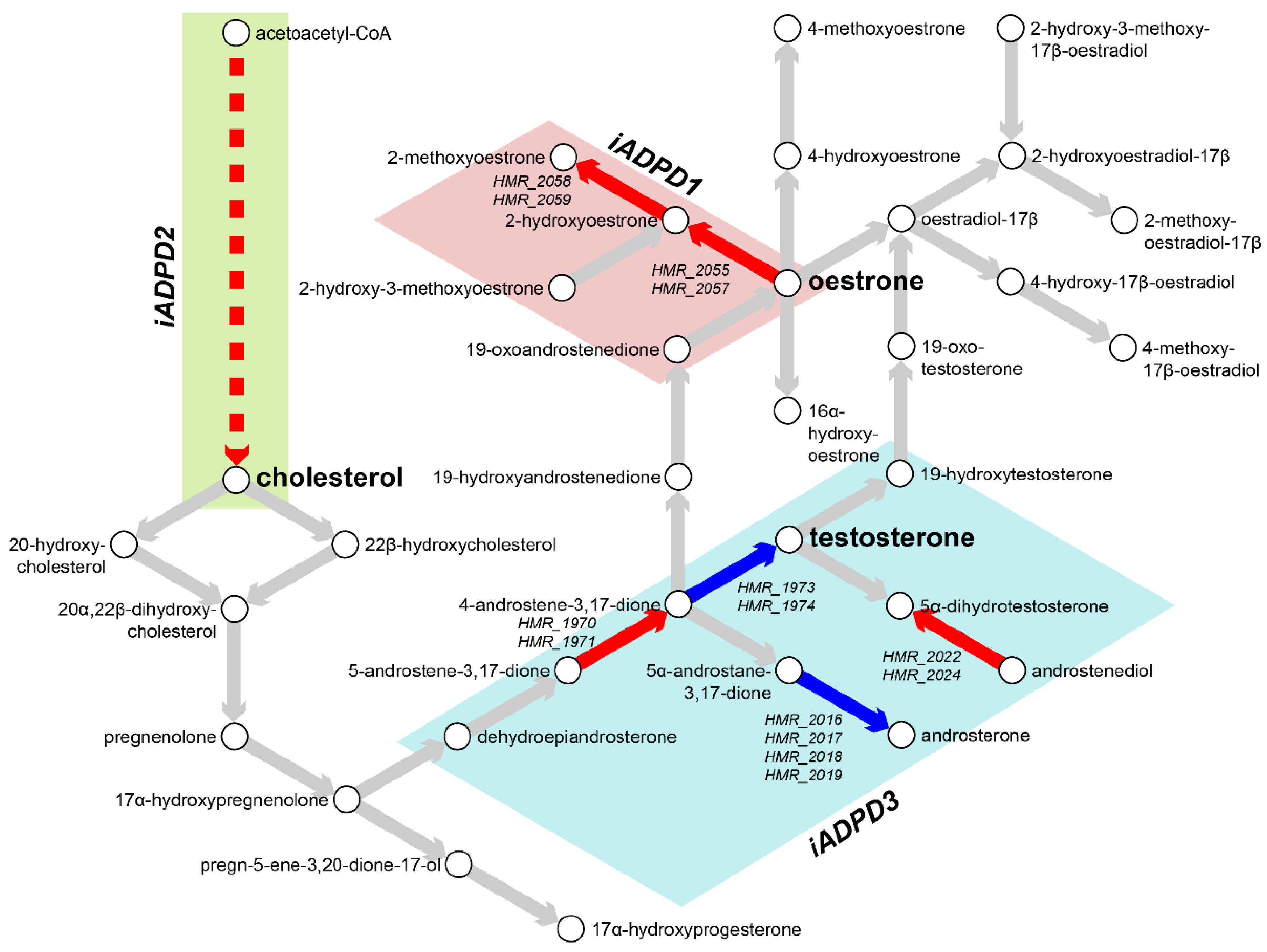
3.2. Metabolic Analysis Revealed Retinoids and Sex Hormones as Significantly Dysregulated in AD and PD
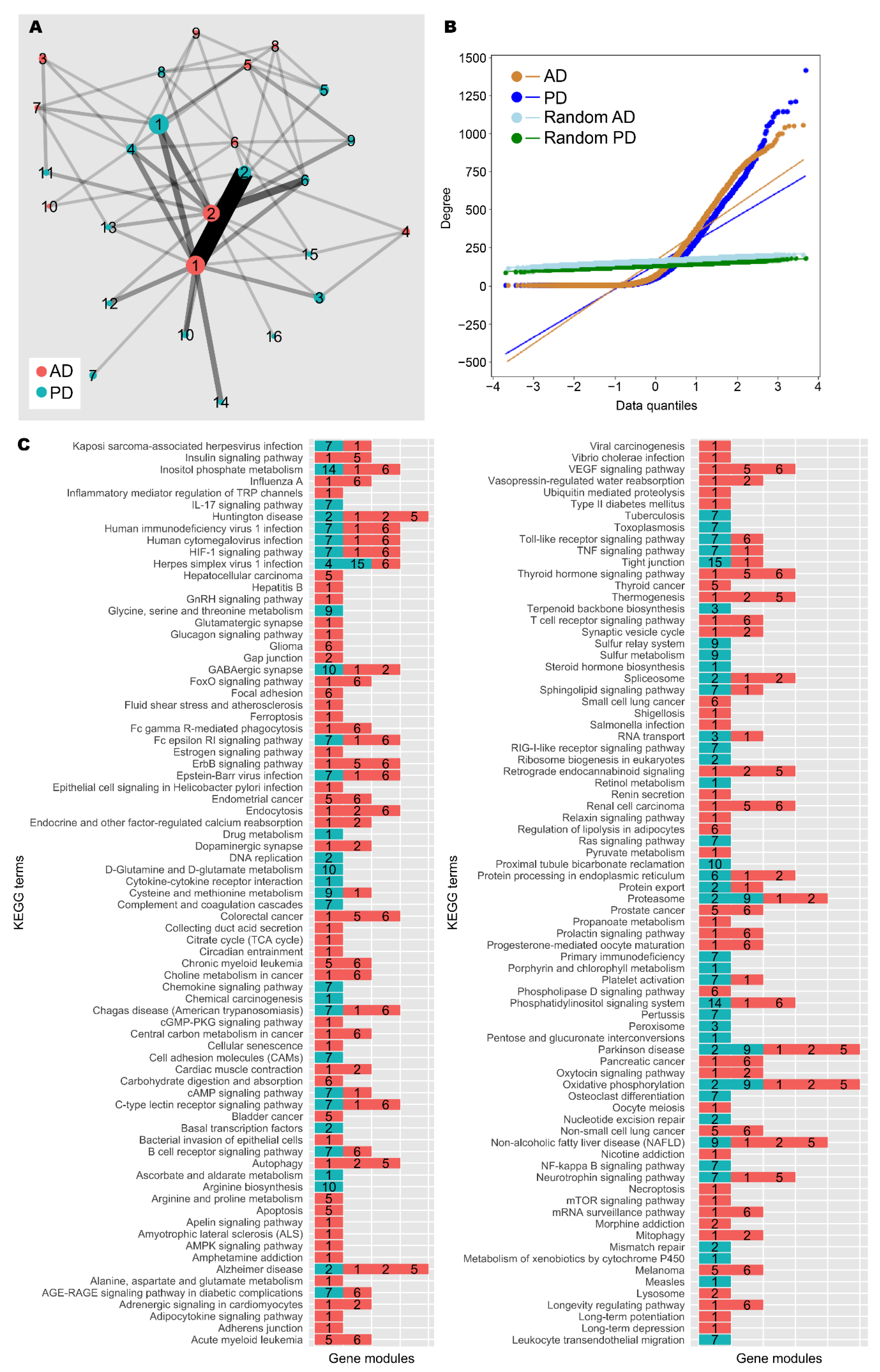
3.3. Network Analysis Supported Retinoid and Androgen Dysregulation and Suggests Transcriptomic Similarity between AD and PD
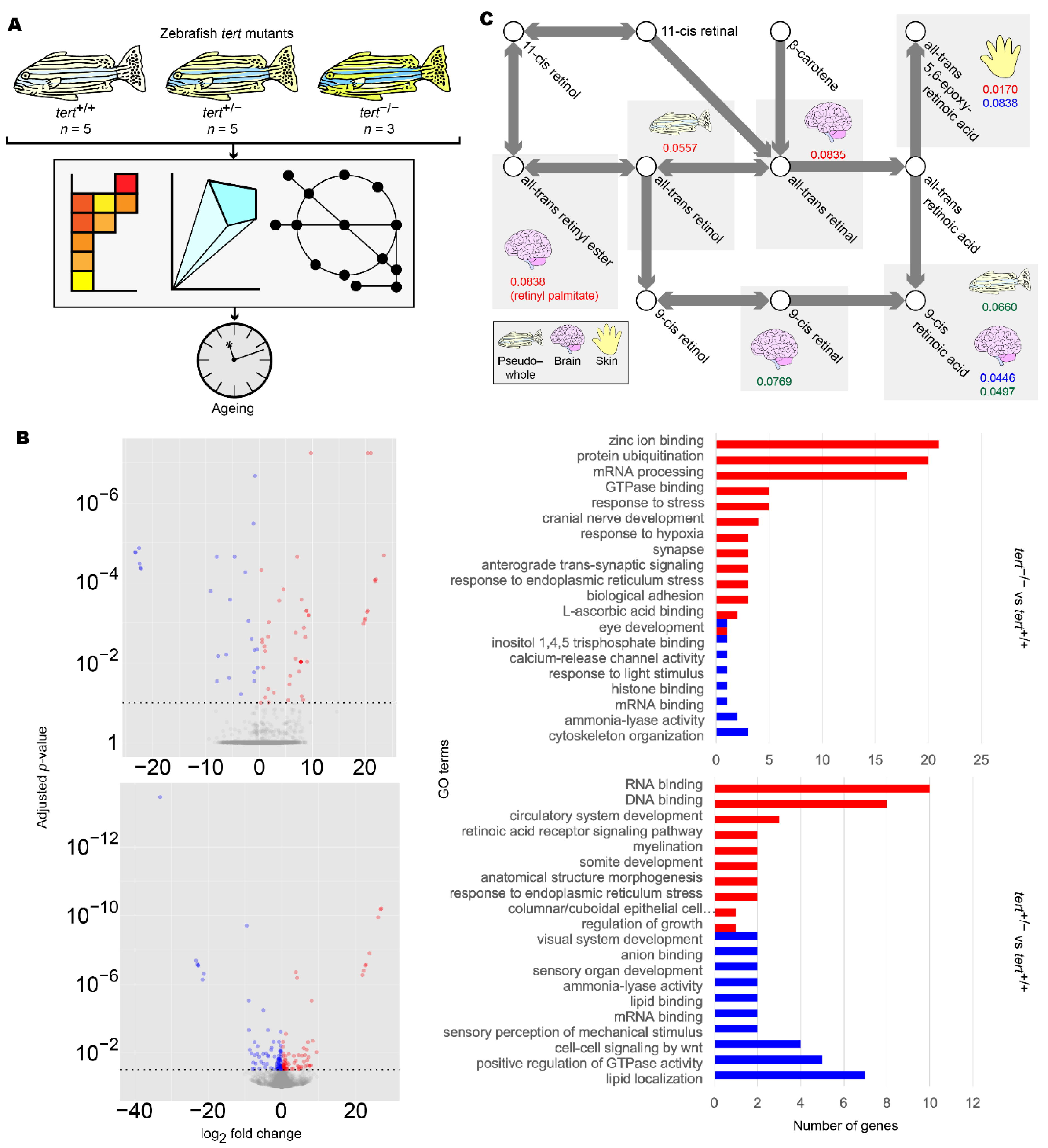
3.4. Zebrafish Transcriptomic and Metabolic Investigations Suggest an Association between Brain Ageing and Retinoid Dysregulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Liberini, P.; Valerio, A.; Memo, M.; Spano, P.F. Lewy-Body Dementia and Responsiveness to Cholinesterase Inhibitors: A Paradigm for Heterogeneity of Alzheimer’s Disease? Trends Pharmacol. Sci. 1996, 17, 155–160. [Google Scholar] [CrossRef]
- Wijemanne, S.; Jankovic, J. Dopa-Responsive Dystonia—Clinical and Genetic Heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Greenland, J.C.; Williams-Gray, C.H.; Barker, R.A. The Clinical Heterogeneity of Parkinson’s Disease and Its Therapeutic Implications. Eur. J. Neurosci. 2019, 49, 328–338. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Baum, L.W. Sex, Hormones, and Alzheimer’s Disease. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2005, 60, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meoni, S.; Macerollo, A.; Moro, E. Sex Differences in Movement Disorders. Nat. Rev. Neurol. 2020, 16, 84–96. [Google Scholar] [CrossRef]
- Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Cholesterol Level and Statin Use in Alzheimer Disease: I. Review of Epidemiological and Preclinical Studies. Arch. Neurol. 2011, 68, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.M.; Jang, W.; Shin, D.W. Association of Statin Use with Parkinson’s Disease: Dose–Response Relationship. Mov. Disord. 2019, 34, 1014–1021. [Google Scholar] [CrossRef]
- Lam, S.; Bayraktar, A.; Zhang, C.; Turkez, H.; Nielsen, J.; Boren, J.; Shoaie, S.; Uhlen, M.; Mardinoglu, A. A Systems Biology Approach for Studying Neurodegenerative Diseases. Drug Discov. Today 2020, 25, 1146–1159. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Boren, J.; Smith, U.; Uhlen, M.; Nielsen, J. Systems Biology in Hepatology: Approaches and Applications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 365–377. [Google Scholar] [CrossRef]
- Lam, S.; Doran, S.; Yuksel, H.H.; Altay, O.; Turkez, H.; Nielsen, J.; Boren, J.; Uhlen, M.; Mardinoglu, A. Addressing the Heterogeneity in Liver Diseases Using Biological Networks. Brief. Bioinform. 2021, 22, 1751–1766. [Google Scholar] [CrossRef]
- Altay, O.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Systems Biology Perspective for Studying the Gut Microbiota in Human Physiology and Liver Diseases. EBioMedicine 2019, 49, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems Biology in Cardiovascular Disease: A Multiomics Approach. Nat. Rev. Cardiol. 2020, 18, 313–330. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Stampfer, M.J. Cardiovascular Disease and Alzheimer’s Disease: Common Links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse Association between Cancer and Alzheimer’s Disease: Results from the Framingham Heart Study. BMJ (Online) 2012, 344, e1442. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, A.; Driver, J.A.; Schernhammer, E.S. Parkinson’s Disease and Cancer Risk: A Systematic Review and Meta-Analysis. Cancer Causes Control 2010, 21, 697–707. [Google Scholar] [CrossRef]
- Sengoku, R. Aging and Alzheimer’s Disease Pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef]
- Hindle, J.V. Ageing, Neurodegeneration and Parkinson’s Disease. Age Ageing 2010, 39, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, Health, and Hallmarks of Aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Carneiro, M.C.; De Castro, I.P.; Ferreira, M.G. Telomeres in Aging and Disease: Lessons from Zebrafish. DMM Dis. Models Mech. 2016, 9, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Anchelin, M.; Alcaraz-Pérez, F.; Martínez, C.M.; Bernabé-García, M.; Mulero, V.; Cayuela, M.L. Premature Aging in Telomerase-Deficient Zebrafish. DMM Dis. Models Mech. 2013, 6, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, C.M.; Carneiro, M.C.; Tenente, I.M.; Jacinto, A.; Ferreira, M.G. Telomerase Is Required for Zebrafish Lifespan. PLoS Genet. 2013, 9, 1003214. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. GTEx Consortium The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; De Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 Promoter Level Mammalian Expression Atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; De Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM Web Resource: Expansion to Provide Additional Transcriptome Atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [Green Version]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-Specific Regulatory Circuits Reveal Variable Modular Perturbations across Complex Diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A Reference Map of the Human Protein Interactome. bioRxiv 2019. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.U.; Patrick, E.; Komashko, V.; et al. A Molecular Network of the Aging Human Brain Provides Insights into the Pathology and Cognitive Decline of Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Myers, A.J.; Gibbs, J.R.; Webster, J.A.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; Leung, D.; Bryden, L.; Nath, P.; et al. A Survey of Genetic Human Cortical Gene Expression. Nat. Genet. 2007, 39, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.A.; Gibbs, J.R.; Clarke, J.; Ray, M.; Zhang, W.; Holmans, P.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; et al. Genetic Control of Human Brain Transcript Expression in Alzheimer Disease. Am. J. Hum. Genet. 2009, 84, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, A.P.; Bidkhori, G.; Shoaie, S.; Clarke, E.; Morrin, H.; Hye, A.; Williams, G.; Ballard, C.; Francis, P.; Aarsland, D. Postmortem Cortical Transcriptomics of Lewy Body Dementia Reveal Mitochondrial Dysfunction and Lack of Neuroinflammation. Am. J. Geriatr. Psychiatry 2020, 28, 75–86. [Google Scholar] [CrossRef]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional Analysis of Multiple Brain Regions in Parkinson’s Disease Supports the Involvement of Specific Protein Processing, Energy Metabolism, and Signaling Pathways, and Suggests Novel Disease Mechanisms. Am. J. Med. Genet. Neuropsychiatr. Genet. 2005, 137 B, 5–16. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Van Der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2625. [Google Scholar]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729.e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the Gene Set Analysis of Genome-Wide Data by Incorporating Directionality of Gene Expression and Combining Statistical Hypotheses and Methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of Clinical Data with a Genome-Scale Metabolic Model of the Human Adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-Scale Metabolic Modelling of Hepatocytes Reveals Serine Deficiency in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A Versatile Toolbox for Metabolic Network Reconstruction and a Case Study on Streptomyces Coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of Genome-Scale Active Metabolic Networks for 69 Human Cell Types and 16 Cancer Types Using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of Anticancer Drugs for Hepatocellular Carcinoma through Personalized Genome-Scale Metabolic Modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef]
- Patil, K.R.; Nielsen, J. Uncovering Transcriptional Regulation of Metabolism by Using Metabolic Network Topology. Proc. Natl. Acad. Sci. USA 2005, 102, 2685–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Aramillo Irizar, P.; Schäuble, S.; Esser, D.; Groth, M.; Frahm, C.; Priebe, S.; Baumgart, M.; Hartmann, N.; Marthandan, S.; Menzel, U.; et al. Transcriptomic Alterations during Ageing Reflect the Shift from Cancer to Degenerative Diseases in the Elderly. Nat. Commun. 2018, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Rajsombath, M.M.; Nam, A.Y.; Ericsson, M.; Nuber, S. Female Sex and Brain-Selective Estrogen Benefit α-Synuclein Tetramerization and the PD-like Motor Syndrome in 3K Transgenic Mice. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 7628–7640. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.M.; Matsumoto, A.M.; Stephens-Shields, A.J.; Ellenberg, S.S.; Gill, T.M.; Shumaker, S.A.; Pleasants, D.D.; Barrett-Connor, E.; Bhasin, S.; Cauley, J.A.; et al. Testosterone Treatment and Cognitive Function in Older Men with Low Testosterone and Age-Associated Memory Impairment. JAMA J. Am. Med. Assoc. 2017, 317, 717–727. [Google Scholar] [CrossRef]
- Wahjoepramono, E.J.; Asih, P.R.; Aniwiyanti, V.; Taddei, K.; Dhaliwal, S.S.; Fuller, S.J.; Foster, J.; Carruthers, M.; Verdile, G.; Sohrabi, H.R.; et al. The Effects of Testosterone Supplementation on Cognitive Functioning in Older Men. CNS Neurol. Disord. Drug Targets 2016, 15, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Pâquet, S.; Caron, P.; Fazli, L.; Rennie, P.S.; Bélanger, A.; Barbier, O. Androgen Glucuronidation: An Unexpected Target for Androgen Deprivation Therapy, with Prognosis and Diagnostic Implications. Cancer Res. 2013, 73, 6963–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomhoff, R.; Blomhoff, H.K. Overview of Retinoid Metabolism and Function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Van Steijn, L.; Verbeek, F.J.; Spaink, H.P.; Merks, R.M.H. Predicting Metabolism from Gene Expression in an Improved Whole-Genome Metabolic Network Model of Danio Rerio. Zebrafish 2019, 16, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Riahi, R.R.; Bush, A.E.; Cohen, P.R. Topical Retinoids: Therapeutic Mechanisms in the Treatment of Photodamaged Skin. Am. J. Clin. Dermatol. 2016, 17, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Dasgupta, S.; Ray, S. Potential Therapeutic Roles of Retinoids for Prevention of Neuroinflammation and Neurodegeneration in Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 1880–1892. [Google Scholar] [PubMed]
- Shudo, K.; Fukasawa, H.; Nakagomi, M.; Yamagata, N. Towards Retinoid Therapy for Alzheimers Disease. Curr. Alzheimer Res. 2009, 6, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A Candidate Retinoid Drug for Alzheimer’s Disease. Biol. Pharm. Bull. 2012, 35, 1206–1212. [Google Scholar] [CrossRef] [Green Version]
- Fitz, N.F.; Nam, K.N.; Koldamova, R.; Lefterov, I. Therapeutic Targeting of Nuclear Receptors, Liver X and Retinoid X Receptors, for Alzheimer’s Disease. Br. J. Pharmacol. 2019, 176, 3599–3610. [Google Scholar] [CrossRef]
- Sinclair, E.; Trivedi, D.K.; Sarkar, D.; Walton-Doyle, C.; Milne, J.; Kunath, T.; Rijs, A.M.; de Bie, R.M.A.; Goodacre, R.; Silverdale, M.; et al. Metabolomics of Sebum Reveals Lipid Dysregulation in Parkinson’s Disease. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Martucci, C.P. The Role of 2-Methoxyestrone in Estrogen Action. J. Steroid Biochem. 1983, 19, 635–638. [Google Scholar] [CrossRef]
- Santana-Santana, M.; Bayascas, J.-R.; Giménez-Llort, L. Fine-Tuning the PI3K/Akt Signaling Pathway Intensity by Sex and Genotype-Load: Sex-Dependent Homozygotic Threshold for Somatic Growth but Feminization of Anxious Phenotype in Middle-Aged PDK1 K465E Knock-In and Heterozygous Mice. Biomedicines 2021, 9, 747. [Google Scholar] [CrossRef] [PubMed]
- Santana-Santana, M.; Bayascas, J.-R.; Giménez-Llort, L. Sex-Dependent Signatures, Time Frames and Longitudinal Fine-Tuning of the Marble Burying Test in Normal and AD-Pathological Aging Mice. Biomedicines 2021, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Muntsant, A.; Jiménez-Altayó, F.; Puertas-Umbert, L.; Jiménez-Xarrie, E.; Vila, E.; Giménez-Llort, L. Sex-Dependent End-of-Life Mental and Vascular Scenarios for Compensatory Mechanisms in Mice with Normal and AD-Neurodegenerative Aging. Biomedicines 2021, 9, 111. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | AD Samples | PD Samples | Control Samples |
|---|---|---|---|
| GTEx/FANTOM5 | 0 | 0 | 67 |
| HPA | 0 | 0 | 52 |
| Rajkumar | 0 | 14 | 13 |
| ROSMAP | 629 | 0 | 704 |
| Zhang/Zheng | 0 | 40 | 53 |
| Total | 629 | 54 | 889 |
| Subsystem | iADPD1 | iADPD2 | iADPD3 |
|---|---|---|---|
| Acyl-CoA hydrolysis | −0.001 | 0.001 | 0.000 |
| Alanine, aspartate, and glutamate metabolism | −0.148 | 0.014 | 0.000 |
| Aminoacyl-tRNA biosynthesis | 4.698 | 4.698 | 0.000 |
| Androgen metabolism | −1.426 | −0.399 | −0.001 |
| Arachidonic acid metabolism | −0.098 | 0.010 | 0.000 |
| Arginine and proline metabolism | −0.182 | −0.327 | 0.000 |
| Beta oxidation of branched-chain fatty acids (mitochondrial) | −0.049 | −0.049 | −0.049 |
| Beta oxidation of di-unsaturated fatty acids (n-6) (mitochondrial) | −0.636 | 0.002 | −0.001 |
| Beta oxidation of odd-chain fatty acids (mitochondrial) | 0.001 | −0.002 | −0.002 |
| Beta oxidation of poly-unsaturated fatty acids (mitochondrial) | 0.709 | 0.024 | 0.000 |
| Beta oxidation of unsaturated fatty acids (n-7) (mitochondrial) | −0.016 | 0.001 | −0.003 |
| Beta oxidation of unsaturated fatty acids (n-9) (mitochondrial) | 0.011 | 0.000 | 0.007 |
| Carnitine shuttle (cytosolic) | 0.012 | 0.000 | −0.001 |
| Carnitine shuttle (mitochondrial) | 0.003 | 0.000 | 0.002 |
| Cholesterol biosynthesis 1 (Bloch pathway) | 0.076 | −0.983 | 0.001 |
| Cholesterol biosynthesis 2 | 2.501 | 4.472 | 0.000 |
| Cholesterol biosynthesis 3 (Kandustch–Russell pathway) | 1.699 | 0.000 | 0.000 |
| Cholesterol metabolism | 0.067 | 4.482 | 0.000 |
| Estrogen metabolism | 2.085 | 0.000 | 0.000 |
| Fatty acid activation (endoplasmic reticular) | 0.000 | 0.000 | 0.000 |
| Fatty acid biosynthesis (even-chain) | 0.000 | 0.000 | 0.000 |
| Fatty acid desaturation (even-chain) | 0.785 | 0.000 | 0.000 |
| Fatty acid elongation (odd-chain) | −0.042 | −0.024 | 0.000 |
| Formation and hydrolysis of cholesterol esters | −0.382 | 0.004 | 0.000 |
| Fructose and mannose metabolism | −0.211 | −0.007 | 0.000 |
| Galactose metabolism | −0.008 | 0.035 | 0.000 |
| Glycine, serine, and threonine metabolism | 0.276 | 0.557 | 0.000 |
| Glycolysis/gluconeogenesis | −0.213 | 0.022 | 0.033 |
| Histidine metabolism | 0.000 | 0.000 | 0.000 |
| Leukotriene metabolism | −0.032 | 0.000 | 0.000 |
| Lysine metabolism | 0.000 | 0.000 | 0.000 |
| N-glycan metabolism | −0.784 | 0.016 | 0.000 |
| Nitrogen metabolism | 0.000 | 0.000 | 0.000 |
| Nucleotide metabolism | 0.027 | −0.028 | 0.000 |
| O-glycan metabolism | −2.346 | −4.738 | 0.000 |
| Pentose phosphate pathway | 0.127 | 0.000 | 0.000 |
| Propanoate metabolism | −0.116 | 0.020 | 0.091 |
| Protein degradation | 0.000 | 0.000 | 0.000 |
| Purine metabolism | 0.112 | −0.013 | 0.000 |
| Pyrimidine metabolism | −0.071 | −0.010 | −0.001 |
| Pyruvate metabolism | −0.183 | −0.004 | −0.077 |
| Starch and sucrose metabolism | 0.000 | 0.000 | 0.000 |
| Steroid metabolism | −0.097 | −0.295 | 0.003 |
| Terpenoid backbone biosynthesis | 0.398 | 0.187 | 0.020 |
| Valine, leucine, and isoleucine degradation | 0.127 | 0.000 | 0.000 |
| Reporter Metabolite | Z-Score | p-Value |
|---|---|---|
| Cluster 1 | ||
| O2 | 6.111 | 4.95 × 10−10 |
| Estrone | 5.4557 | 2.44 × 10−8 |
| Retinoate | 5.3943 | 3.44 × 10−8 |
| NADP+ | 5.3667 | 4.01 × 10−8 |
| Arachidonate | 5.2822 | 6.38 × 10−8 |
| 2-Hydroxyestradiol-17beta | 5.0999 | 1.70 × 10−7 |
| Linoleate | 5.0622 | 2.07 × 10−7 |
| 10-HETE | 5.0454 | 2.26 × 10−7 |
| 11,12,15-THETA | 5.0454 | 2.26 × 10−7 |
| 11,14,15-Theta | 5.0454 | 2.26 × 10−7 |
| Cluster 2 | ||
| 1-Acylglycerol-3P-LD-PC pool | 4.3322 | 7.38 × 10−6 |
| Acyl-CoA-LD-PI pool | 4.143 | 1.71 × 10−5 |
| Phosphatidate-CL pool | 4.0973 | 2.09 × 10−5 |
| Thymidine | 3.5852 | 0.00016843 |
| Uridine | 3.5852 | 0.00016843 |
| Prostaglandin D2 | 3.2144 | 0.00065348 |
| G10596 | 3.1354 | 0.0008581 |
| G10597 | 3.1354 | 0.0008581 |
| D-Myo-inositol-1,4,5-trisphosphate | 2.9988 | 0.0013552 |
| Dolichyl-phosphate | 2.9655 | 0.001511 |
| Cluster 3 | ||
| D-Myo-inositol-1,4,5-trisphosphate | 2.6543 | 0.0039734 |
| 13-cis-Retinal | 2.6537 | 0.0039806 |
| Heparan sulfate, precursor 9 | 2.5915 | 0.0047772 |
| sn-Glycerol-3-phosphate | 2.578 | 0.0049682 |
| DHAP | 2.5353 | 0.0056173 |
| Porphobilinogen | 2.4987 | 0.0062333 |
| ATP | 2.4838 | 0.0064998 |
| L-Glutamate 5-semialdehyde | 2.4576 | 0.006994 |
| Prostaglandin D2 | 2.451 | 0.0071221 |
| ribose | 2.4133 | 0.0079045 |
| Nodes | Edges | Diameter | Average Path Length | Density | Clustering Coefficient | Connected Network? | Minimum Cut | |
|---|---|---|---|---|---|---|---|---|
| AD | 4861 | 396,985 | 11 | 3.004 | 0.034 | 0.443 | No | - |
| PD | 5857 | 394,405 | 18 | 3.598 | 0.023 | 0.397 | No | - |
| Random AD | 4861 | 396,985 | 3 | 1.970 | 0.034 | 0.034 | Yes | 114 |
| Random PD | 5857 | 394,405 | 3 | 2.021 | 0.023 | 0.023 | Yes | 89 |
| Reporter Metabolite | Z-Score | p-Value |
|---|---|---|
| tert−/− | ||
| H+ | 3.911 | 4.60 × 10−5 |
| H2O | 3.0672 | 0.0010804 |
| L-Lysine | 2.8564 | 0.0021424 |
| Biocyt c | 2.8564 | 0.0021424 |
| Ubiquinone | 2.5742 | 0.0050241 |
| Nicotinamide adenine dinucleotide—reduced | 2.3946 | 0.0083183 |
| Phosphate | 2.0562 | 0.019883 |
| Superoxide anion | 2.0365 | 0.020851 |
| Sodium | 1.9228 | 0.027254 |
| TRNA (Glu) | 1.8752 | 0.030381 |
| Thiosulfate | 1.7684 | 0.038493 |
| Selenate | 1.7684 | 0.038493 |
| Reduced glutathione | 1.7184 | 0.042862 |
| ADP | 1.6716 | 0.047305 |
| L-Lysine | 1.6625 | 0.04821 |
| Benzo[a]pyrene-4,5-oxide | 1.6042 | 0.054333 |
| Formaldehyde | 1.5955 | 0.055302 |
| L-Glutamate | 1.4622 | 0.071837 |
| (1R,2S)-Naphthalene epoxide | 1.4518 | 0.073276 |
| Aflatoxin B1 exo-8,9-epozide | 1.4518 | 0.073276 |
| tert+/− | ||
| H+ | 4.9585 | 3.55 × 10−7 |
| Ubiquinol | 3.9938 | 3.25 × 10−5 |
| H2O | 3.2078 | 0.00066883 |
| Nicotinamide adenine dinucleotide—reduced | 3.029 | 0.0012268 |
| Superoxide anion | 2.0908 | 0.018274 |
| L-Lactate | 2.0752 | 0.018983 |
| O2 | 1.9958 | 0.022976 |
| Lnlncgcoa c | 1.9628 | 0.024834 |
| Succinate | 1.9449 | 0.025895 |
| Ferricytochrome c | 1.8352 | 0.033237 |
| Phosphatidylinositol-3,4,5-trisphosphate | 1.7494 | 0.040109 |
| 9-cis-Retinoic acid | 1.7 | 0.044567 |
| [(Gal)2 (GlcNAc)4 (LFuc)1 (Man)3 (Asn)1’] | 1.6672 | 0.047739 |
| O-Phospho-L-serine | 1.6601 | 0.048451 |
| [(Glc)3 (GlcNAc)2 (Man)9 (Asn)1’] | 1.6276 | 0.051802 |
| Protein serine | 1.6078 | 0.053937 |
| [(GlcNAc)1 (Ser/Thr)1’] | 1.6078 | 0.053937 |
| Geranyl diphosphate | 1.5912 | 0.055785 |
| CTP | 1.5625 | 0.059088 |
| [(Gal)2 (GlcNAc)4 (LFuc)1 (Man)3 (Neu5Ac)2 (Asn)1’] | 1.5367 | 0.062179 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, S.; Hartmann, N.; Benfeitas, R.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Englert, C.; Knight, R.; Mardinoglu, A. Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines 2021, 9, 1310. https://doi.org/10.3390/biomedicines9101310
Lam S, Hartmann N, Benfeitas R, Zhang C, Arif M, Turkez H, Uhlén M, Englert C, Knight R, Mardinoglu A. Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines. 2021; 9(10):1310. https://doi.org/10.3390/biomedicines9101310
Chicago/Turabian StyleLam, Simon, Nils Hartmann, Rui Benfeitas, Cheng Zhang, Muhammad Arif, Hasan Turkez, Mathias Uhlén, Christoph Englert, Robert Knight, and Adil Mardinoglu. 2021. "Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases" Biomedicines 9, no. 10: 1310. https://doi.org/10.3390/biomedicines9101310
APA StyleLam, S., Hartmann, N., Benfeitas, R., Zhang, C., Arif, M., Turkez, H., Uhlén, M., Englert, C., Knight, R., & Mardinoglu, A. (2021). Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines, 9(10), 1310. https://doi.org/10.3390/biomedicines9101310