
Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Obesity



 and
and
Abstract
:1. Introduction
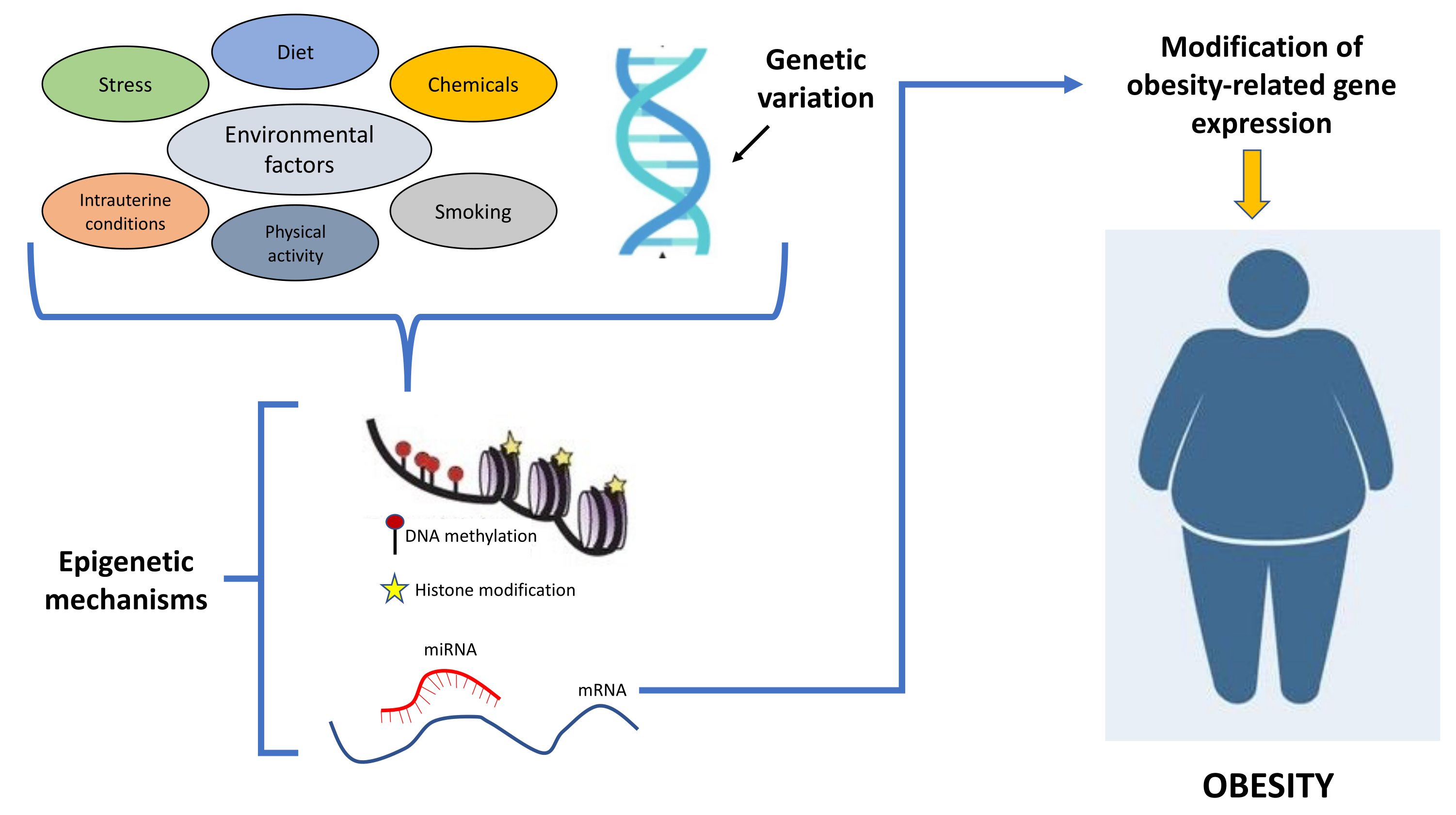
2. Epigenetics and Obesity
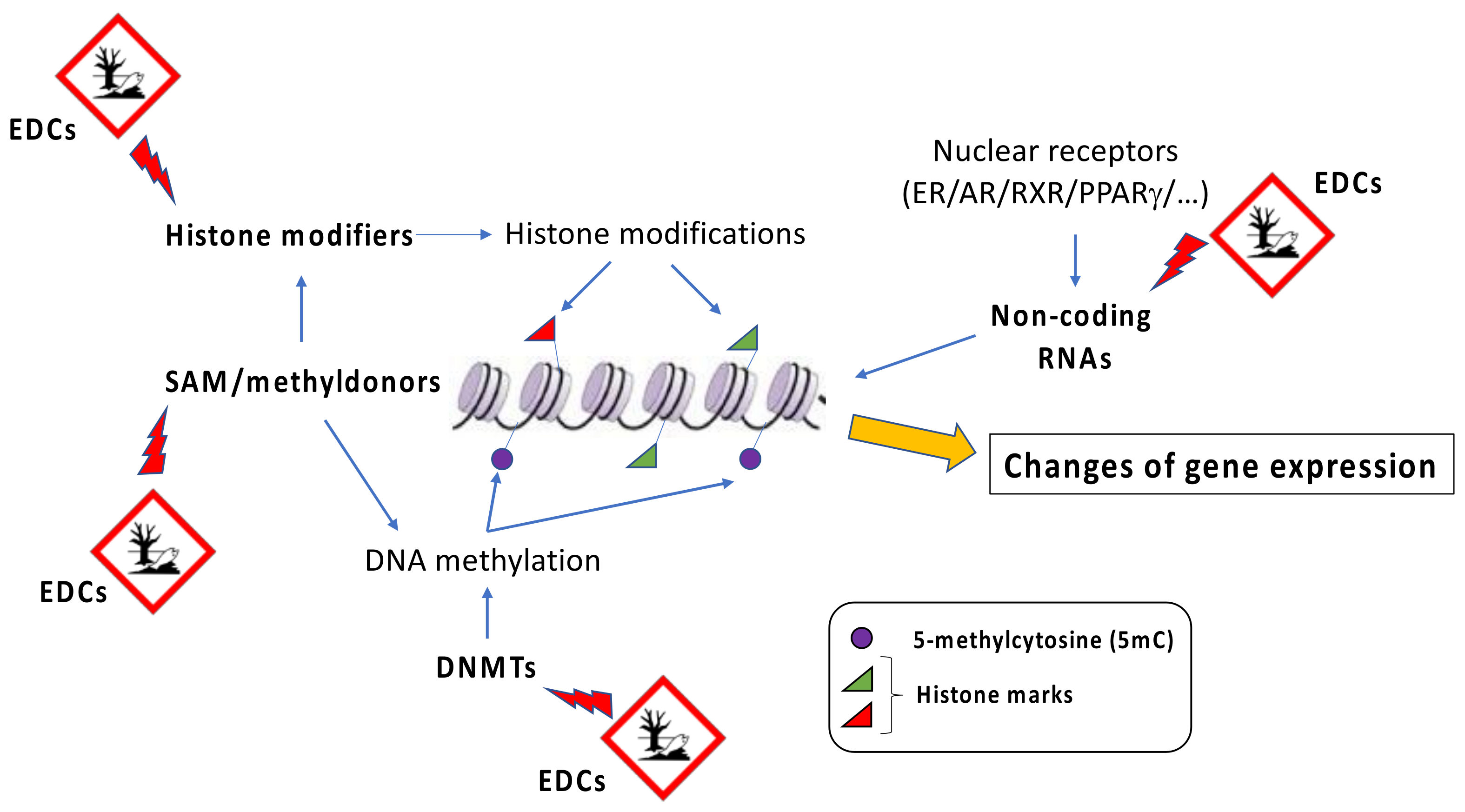
3. Epigenetic Changes Induced by Obesogenic EDCs Exposure
3.1. Bisphenol A
3.2. Diethylstilbestrol
3.3. Phthalates
3.4. Organochlorine and Organophosphate Pesticides
3.5. Inhaled Pollutants
3.6. Flame Retardants
4. Epigenetic Inheritance Determined by Obesogenic EDCs
5. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Macchia, P.E.; Nettore, I.C.; Franchini, F.; Santana-Viera, L.; Ungaro, P. Epigenetic regulation of adipogenesis by histone-modifying enzymes. Epigenomics 2021, 13, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Hampton, T. Scientists study fat as endocrine organ. JAMA 2006, 296, 1573–1575. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulou, L.; Psycharakis, C.; Petrakis, D.; Tsiaoussis, J.; Tsatsakis, A.M. Obesity, Persistent Organic Pollutants and Related Health Problems. Adv. Exp. Med. Biol. 2017, 960, 81–110. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, M.; Llewellyn, A.; Owen, C.G.; Woolacott, N. Predicting adult obesity from childhood obesity: A systematic review and meta-analysis. Obes. Rev. 2016, 17, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, A.; Oriente, F.; Iovino, S.; Alberobello, A.T.; Barbagallo, A.P.; Esposito, I.; Fiory, F.; Teperino, R.; Ungaro, P.; Miele, C.; et al. Phorbol esters induce intracellular accumulation of the anti-apoptotic protein PED/PEA-15 by preventing ubiquitinylation and proteasomal degradation. J. Biol. Chem. 2007, 282, 8648–8657. [Google Scholar] [CrossRef] [Green Version]
- Willinger, L.; Brudy, L.; Meyer, M.; Oberhoffer-Fritz, R.; Ewert, P.; Muller, J. Overweight and Obesity in Patients with Congenital Heart Disease: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 9931. [Google Scholar] [CrossRef]
- Swinburn, B.; Egger, G. Preventive strategies against weight gain and obesity. Obes. Rev. 2002, 3, 289–301. [Google Scholar] [CrossRef]
- Kabir, E.R.; Rahman, M.S.; Rahman, I. A review on endocrine disruptors and their possible impacts on human health. Environ. Toxicol. Pharmacol. 2015, 40, 241–258. [Google Scholar] [CrossRef]
- Nettore, I.C.; Colao, A.; Macchia, P.E. Nutritional and Environmental Factors in Thyroid Carcinogenesis. Int. J. Environ. Res. Public Health 2018, 15, 1735. [Google Scholar] [CrossRef] [Green Version]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 2016, 130, 943–986. [Google Scholar] [CrossRef] [Green Version]
- Ollikainen, M.; Ismail, K.; Gervin, K.; Kyllonen, A.; Hakkarainen, A.; Lundbom, J.; Jarvinen, E.A.; Harris, J.R.; Lundbom, N.; Rissanen, A.; et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenetics 2015, 7, 39. [Google Scholar] [CrossRef]
- Demerath, E.W.; Guan, W.; Grove, M.L.; Aslibekyan, S.; Mendelson, M.; Zhou, Y.H.; Hedman, A.K.; Sandling, J.K.; Li, L.A.; Irvin, M.R.; et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 2015, 24, 4464–4479. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.L.; Vastolo, V.; Ciccarelli, M.; Albano, L.; Macchia, P.E.; Ungaro, P. Dietary polyphenols and chromatin remodeling. Crit. Rev. Food Sci. Nutr. 2017, 57, 2589–2599. [Google Scholar] [CrossRef]
- Petronis, A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010, 465, 721–727. [Google Scholar] [CrossRef]
- Nettore, I.C.; Rocca, C.; Mancino, G.; Albano, L.; Amelio, D.; Grande, F.; Puoci, F.; Pasqua, T.; Desiderio, S.; Mazza, R.; et al. Quercetin and its derivative Q2 modulate chromatin dynamics in adipogenesis and Q2 prevents obesity and metabolic disorders in rats. J. Nutr. Biochem. 2019, 69, 151–162. [Google Scholar] [CrossRef]
- Lima, R.S.; de Assis Silva Gomes, J.; Moreira, P.R. An overview about DNA methylation in childhood obesity: Characteristics of the studies and main findings. J. Cell. Biochem. 2020, 121, 3042–3057. [Google Scholar] [CrossRef]
- Raciti, G.A.; Longo, M.; Parrillo, L.; Ciccarelli, M.; Mirra, P.; Ungaro, P.; Formisano, P.; Miele, C.; Beguinot, F. Understanding type 2 diabetes: From genetics to epigenetics. Acta Diabetol. 2015, 52, 821–827. [Google Scholar] [CrossRef]
- Porta, M.; Amione, C.; Barutta, F.; Fornengo, P.; Merlo, S.; Gruden, G.; Albano, L.; Ciccarelli, M.; Ungaro, P.; Durazzo, M.; et al. The co-activator-associated arginine methyltransferase 1 (CARM1) gene is overexpressed in type 2 diabetes. Endocrine 2019, 63, 284–292. [Google Scholar] [CrossRef]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Castel, S.E.; Martienssen, R.A. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013, 14, 100–112. [Google Scholar] [CrossRef]
- Petryk, N.; Bultmann, S.; Bartke, T.; Defossez, P.A. Staying true to yourself: Mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res. 2021, 49, 3020–3032. [Google Scholar] [CrossRef]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596. [Google Scholar] [CrossRef]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef]
- Capparelli, R.; Iannelli, D. Role of Epigenetics in Type 2 Diabetes and Obesity. Biomedicines 2021, 9, 977. [Google Scholar] [CrossRef]
- Vastolo, V.; Nettore, I.C.; Ciccarelli, M.; Albano, L.; Raciti, G.A.; Longo, M.; Beguinot, F.; Ungaro, P. High-fat diet unveils an enhancer element at the Ped/Pea-15 gene responsible for epigenetic memory in skeletal muscle. Metabolism 2018, 87, 70–79. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Vastolo, V.; Albano, L.; Lecce, M.; Cabaro, S.; Liotti, A.; Longo, M.; Oriente, F.; Russo, G.L.; Macchia, P.E.; et al. Glucose-induced expression of the homeotic transcription factor Prep1 is associated with histone post-translational modifications in skeletal muscle. Diabetologia 2016, 59, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.C.; Irvin, M.R.; Claas, S.A.; Arnett, D.K. Lipid Phenotypes and DNA Methylation: A Review of the Literature. Curr. Atheroscler. Rep. 2021, 23, 71. [Google Scholar] [CrossRef]
- Ali, O.; Cerjak, D.; Kent, J.W., Jr.; James, R.; Blangero, J.; Carless, M.A.; Zhang, Y. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics 2016, 11, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannat Ali Pour, N.; Meshkani, R.; Toolabi, K.; Mohassel Azadi, S.; Zand, S.; Emamgholipour, S. Adipose tissue mRNA expression of HDAC1, HDAC3 and HDAC9 in obese women in relation to obesity indices and insulin resistance. Mol. Biol. Rep. 2020, 47, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Jha, T.; Ghosh, B. Dissecting Histone Deacetylase 3 in Multiple Disease Conditions: Selective Inhibition as a Promising Therapeutic Strategy. J. Med. Chem. 2021, 64, 8827–8869. [Google Scholar] [CrossRef]
- Hanzu, F.A.; Musri, M.M.; Sanchez-Herrero, A.; Claret, M.; Esteban, Y.; Kaliman, P.; Gomis, R.; Parrizas, M. Histone demethylase KDM1A represses inflammatory gene expression in preadipocytes. Obesity 2013, 21, E616–E625. [Google Scholar] [CrossRef]
- Duteil, D.; Tosic, M.; Lausecker, F.; Nenseth, H.Z.; Muller, J.M.; Urban, S.; Willmann, D.; Petroll, K.; Messaddeq, N.; Arrigoni, L.; et al. Lsd1 Ablation Triggers Metabolic Reprogramming of Brown Adipose Tissue. Cell Rep. 2016, 17, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Ferland-McCollough, D.; Fernandez-Twinn, D.S.; Cannell, I.G.; David, H.; Warner, M.; Vaag, A.A.; Bork-Jensen, J.; Brons, C.; Gant, T.W.; Willis, A.E.; et al. Programming of adipose tissue miR-483-3p and GDF-3 expression by maternal diet in type 2 diabetes. Cell Death Differ. 2012, 19, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sinnott-Armstrong, N.; Wagschal, A.; Wark, A.R.; Camporez, J.P.; Perry, R.J.; Ji, F.; Sohn, Y.; Oh, J.; Wu, S.; et al. A MicroRNA Linking Human Positive Selection and Metabolic Disorders. Cell 2020, 183, 684–701. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Meng, X.; Sui, X.; Wang, S.; Shen, T.; Huang, X.; Guo, J.; Fang, W.; Man, Y.; Xi, J.; et al. MiR-19a regulates PTEN expression to mediate glycogen synthesis in hepatocytes. Sci. Rep. 2015, 5, 11602. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Liu, C.; Qiao, A.; Cui, Y.; Zhang, H.; Cui, A.; Zhang, S.; Yang, Y.; Xiao, X.; Chen, Y.; et al. MicroRNA-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J. Hepatol. 2013, 58, 535–542. [Google Scholar] [CrossRef]
- Zhuo, S.; Yang, M.; Zhao, Y.; Chen, X.; Zhang, F.; Li, N.; Yao, P.; Zhu, T.; Mei, H.; Wang, S.; et al. MicroRNA-451 Negatively Regulates Hepatic Glucose Production and Glucose Homeostasis by Targeting Glycerol Kinase-Mediated Gluconeogenesis. Diabetes 2016, 65, 3276–3288. [Google Scholar] [CrossRef] [Green Version]
- Le Magueresse-Battistoni, B.; Labaronne, E.; Vidal, H.; Naville, D. Endocrine disrupting chemicals in mixture and obesity, diabetes and related metabolic disorders. World J. Biol. Chem. 2017, 8, 108–119. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef]
- Rhomberg, L.R.; Goodman, J.E. Low-dose effects and nonmonotonic dose-responses of endocrine disrupting chemicals: Has the case been made? Regul. Toxicol. Pharmacol. 2012, 64, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Tabb, M.M.; Blumberg, B. New modes of action for endocrine-disrupting chemicals. Mol. Endocrinol. 2006, 20, 475–482. [Google Scholar] [CrossRef]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Ribeiro, E.; Ladeira, C.; Viegas, S. EDCs Mixtures: A Stealthy Hazard for Human Health? Toxics 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Baillie-Hamilton, P.F. Chemical toxins: A hypothesis to explain the global obesity epidemic. J. Altern. Complement. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef]
- Grun, F.; Blumberg, B. Environmental obesogens: Organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006, 147, S50–S55. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Cheikh Rouhou, M.; Karelis, A.D.; St-Pierre, D.H.; Lamontagne, L. Adverse effects of weight loss: Are persistent organic pollutants a potential culprit? Diabetes Metab. 2016, 42, 215–223. [Google Scholar] [CrossRef]
- Ho, S.M.; Johnson, A.; Tarapore, P.; Janakiram, V.; Zhang, X.; Leung, Y.K. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012, 53, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Anghel, S.I.; Wahli, W. Fat poetry: A kingdom for PPAR gamma. Cell Res. 2007, 17, 486–511. [Google Scholar] [CrossRef] [Green Version]
- Christodoulides, C.; Vidal-Puig, A. PPARs and adipocyte function. Mol. Cell. Endocrinol. 2010, 318, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, P.; Mirra, P.; Oriente, F.; Nigro, C.; Ciccarelli, M.; Vastolo, V.; Longo, M.; Perruolo, G.; Spinelli, R.; Formisano, P.; et al. Peroxisome proliferator-activated receptor-gamma activation enhances insulin-stimulated glucose disposal by reducing ped/pea-15 gene expression in skeletal muscle cells: Evidence for involvement of activator protein-1. J. Biol. Chem. 2012, 287, 42951–42961. [Google Scholar] [CrossRef] [Green Version]
- Grun, F.; Blumberg, B. Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Grun, F.; Blumberg, B. Minireview: The case for obesogens. Mol. Endocrinol. 2009, 23, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Bastos Sales, L.; Kamstra, J.H.; Cenijn, P.H.; van Rijt, L.S.; Hamers, T.; Legler, J. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol. In Vitro 2013, 27, 1634–1643. [Google Scholar] [CrossRef]
- Chevalier, N.; Fenichel, P. Endocrine disruptors: New players in the pathophysiology of type 2 diabetes? Diabetes Metab. 2015, 41, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newbold, R.R.; Padilla-Banks, E.; Snyder, R.J.; Phillips, T.M.; Jefferson, W.N. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod. Toxicol. 2007, 23, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Newbold, R.R. Developmental exposure to endocrine-disrupting chemicals programs for reproductive tract alterations and obesity later in life. Am. J. Clin. Nutr. 2011, 94, 1939S–1942S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Bisphenol-A and diethylstilbestrol exposure induces the expression of breast cancer associated long noncoding RNA HOTAIR in vitro and in vivo. J. Steroid Biochem. Mol. Biol. 2014, 141, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divoux, A.; Karastergiou, K.; Xie, H.; Guo, W.; Perera, R.J.; Fried, S.K.; Smith, S.R. Identification of a novel lncRNA in gluteal adipose tissue and evidence for its positive effect on preadipocyte differentiation. Obesity 2014, 22, 1781–1785. [Google Scholar] [CrossRef] [Green Version]
- Meruvu, S.; Zhang, J.; Choudhury, M. Butyl Benzyl Phthalate Promotes Adipogenesis in 3T3-L1 Cells via the miRNA-34a-5p Signaling Pathway in the Absence of Exogenous Adipogenic Stimuli. Chem. Res. Toxicol. 2021, 34, 2251–2260. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, M.J. Phthalate exposure and childhood obesity. Ann. Pediatr. Endocrinol. Metab. 2014, 19, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Buser, M.C.; Murray, H.E.; Scinicariello, F. Age and sex differences in childhood and adulthood obesity association with phthalates: Analyses of NHANES 2007–2010. Int. J. Hyg. Environ. Health 2014, 217, 687–694. [Google Scholar] [CrossRef]
- Zhang, J.; Choudhury, M. Benzyl Butyl Phthalate Induced Early lncRNA H19 Regulation in C3H10T1/2 Stem Cell Line. Chem. Res. Toxicol. 2021, 34, 54–62. [Google Scholar] [CrossRef]
- Darbre, P. Endocrine Disruption and Human Health, 1st ed.; Darbre, P., Ed.; Academic Press: London, UK, 2015; p. 377. [Google Scholar]
- Slotkin, T.A. Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity? Reprod. Toxicol. 2011, 31, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Orton, F.; Rosivatz, E.; Scholze, M.; Kortenkamp, A. Widely used pesticides with previously unknown endocrine activity revealed as in vitro antiandrogens. Environ. Health Perspect. 2011, 119, 794–800. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, S.; Kieu, T.; Chow, C.; Casey, S.; Blumberg, B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol. Endocrinol. 2010, 24, 526–539. [Google Scholar] [CrossRef]
- Janesick, A.; Blumberg, B. Minireview: PPARgamma as the target of obesogens. J. Steroid Biochem. Mol. Biol. 2011, 127, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Chapman, R.S.; He, X.; Blair, A.E.; Lan, Q. Improvement in household stoves and risk of chronic obstructive pulmonary disease in Xuanwei, China: Retrospective cohort study. BMJ 2005, 331, 1050. [Google Scholar] [CrossRef] [Green Version]
- Hnizdo, E.; Sullivan, P.A.; Bang, K.M.; Wagner, G. Airflow obstruction attributable to work in industry and occupation among U.S. race/ethnic groups: A study of NHANES III data. Am. J. Ind. Med. 2004, 46, 126–135. [Google Scholar] [CrossRef]
- Hiraiwa, K.; van Eeden, S.F. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Zhang, H.; Maher, C.; Arteaga-Solis, E.; Champagne, F.A.; Wu, L.; McDonald, J.D.; Yan, B.; Schwartz, G.J.; Miller, R.L. Prenatal polycyclic aromatic hydrocarbon, adiposity, peroxisome proliferator-activated receptor (PPAR) gamma methylation in offspring, grand-offspring mice. PLoS ONE 2014, 9, e110706. [Google Scholar] [CrossRef] [Green Version]
- Kamstra, J.H.; Hruba, E.; Blumberg, B.; Janesick, A.; Mandrup, S.; Hamers, T.; Legler, J. Transcriptional and epigenetic mechanisms underlying enhanced in vitro adipocyte differentiation by the brominated flame retardant BDE-47. Environ. Sci. Technol. 2014, 48, 4110–4119. [Google Scholar] [CrossRef]
- Halden, R.U. Plastics and health risks. Annu. Rev. Public Health 2010, 31, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Reddivari, L.; Veeramachaneni, D.N.R.; Walters, W.A.; Lozupone, C.; Palmer, J.; Hewage, M.K.K.; Bhatnagar, R.; Amir, A.; Kennett, M.J.; Knight, R.; et al. Perinatal Bisphenol A Exposure Induces Chronic Inflammation in Rabbit Offspring via Modulation of Gut Bacteria and Their Metabolites. mSystems 2017, 2, e00093-17. [Google Scholar] [CrossRef] [Green Version]
- Lakind, J.S.; Goodman, M.; Mattison, D.R. Bisphenol A and indicators of obesity, glucose metabolism/type 2 diabetes and cardiovascular disease: A systematic review of epidemiologic research. Crit. Rev. Toxicol. 2014, 44, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Ranciere, F.; Lyons, J.G.; Loh, V.H.; Botton, J.; Galloway, T.; Wang, T.; Shaw, J.E.; Magliano, D.J. Bisphenol A and the risk of cardiometabolic disorders: A systematic review with meta-analysis of the epidemiological evidence. Environ. Health 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, J.P.; Flachs, E.M.; Rimborg, S.; Glazer, C.H.; Giwercman, A.; Ramlau-Hansen, C.H.; Hougaard, K.S.; Hoyer, B.B.; Haervig, K.K.; Petersen, S.B.; et al. The epidemiologic evidence linking prenatal and postnatal exposure to endocrine disrupting chemicals with male reproductive disorders: A systematic review and meta-analysis. Hum. Reprod. Update 2016, 23, 104–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the mighty and the mutagenic. Toxicol. Rep. 2018, 5, 76–84. [Google Scholar] [CrossRef]
- Richter, C.A.; Birnbaum, L.S.; Farabollini, F.; Newbold, R.R.; Rubin, B.S.; Talsness, C.E.; Vandenbergh, J.G.; Walser-Kuntz, D.R.; vom Saal, F.S. In vivo effects of bisphenol A in laboratory rodent studies. Reprod. Toxicol. 2007, 24, 199–224. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.H.; Pallaroni, L.; Yoon, K.; Gaido, K.; Ross, S.; McDonnell, D. Problems for risk assessment of endocrine-active estrogenic compounds. Environ. Health Perspect. 2002, 110 (Suppl. S6), 925–929. [Google Scholar] [CrossRef] [Green Version]
- Hayes, L.; Weening, A.; Morey, L.M. Differential Effects of Estradiol and Bisphenol A on SET8 and SIRT1 Expression in Ovarian Cancer Cells. Dose Response 2016, 14, 1559325816640682. [Google Scholar] [CrossRef] [Green Version]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef]
- Ben-Jonathan, N.; Hugo, E.R.; Brandebourg, T.D. Effects of bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol. Cell. Endocrinol. 2009, 304, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Carwile, J.L.; Michels, K.B. Urinary bisphenol A and obesity: NHANES 2003-2006. Environ. Res. 2011, 111, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Menale, C.; Grandone, A.; Nicolucci, C.; Cirillo, G.; Crispi, S.; Di Sessa, A.; Marzuillo, P.; Rossi, S.; Mita, D.G.; Perrone, L.; et al. Bisphenol A is associated with insulin resistance and modulates adiponectin and resistin gene expression in obese children. Pediatr. Obes. 2017, 12, 380–387. [Google Scholar] [CrossRef]
- Codoner-Franch, P.; Alonso-Iglesias, E. Resistin: Insulin resistance to malignancy. Clin. Chim. Acta 2015, 438, 46–54. [Google Scholar] [CrossRef]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11beta-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef]
- Boucher, J.G.; Boudreau, A.; Atlas, E. Bisphenol A induces differentiation of human preadipocytes in the absence of glucocorticoid and is inhibited by an estrogen-receptor antagonist. Nutr. Diabetes 2014, 4, e102. [Google Scholar] [CrossRef] [Green Version]
- Hatch, E.E.; Troisi, R.; Palmer, J.R.; Wise, L.A.; Titus, L.; Strohsnitter, W.C.; Ricker, W.; Hyer, M.; Hoover, R.N. Prenatal diethylstilbestrol exposure and risk of obesity in adult women. J. Dev. Orig. Health Dis. 2015, 6, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Gao, W.W.; Tang, H.M.; Deng, J.J.; Wong, C.M.; Chan, C.P.; Jin, D.Y. beta-TrCP-mediated ubiquitination and degradation of liver-enriched transcription factor CREB-H. Sci. Rep. 2016, 6, 23938. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Yang, W.; Ruan, X.; Kiesewetter, K.; Zhu, J.; Cao, H. Integrative Transcriptome Analyses of Metabolic Responses in Mice Define Pivotal LncRNA Metabolic Regulators. Cell Metab. 2016, 24, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Hajjari, M.; Salavaty, A. HOTAIR: An oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 2015, 12, 1–9. [Google Scholar] [CrossRef]
- Calafat, A.M.; McKee, R.H. Integrating biomonitoring exposure data into the risk assessment process: Phthalates [diethyl phthalate and di(2-ethylhexyl) phthalate] as a case study. Environ. Health Perspect. 2006, 114, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.M.; Jones, T.H. Testosterone and obesity. Obes. Rev. 2015, 16, 581–606. [Google Scholar] [CrossRef]
- Svechnikov, K.; Izzo, G.; Landreh, L.; Weisser, J.; Soder, O. Endocrine disruptors and Leydig cell function. J. Biomed. Biotechnol. 2010, 2010, 684504. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Fernandes, A.; Demaegdt, H.; Vandermeiren, K.; Hectors, T.L.; Jorens, P.G.; Blust, R.; Vanparys, C. Evaluation of a screening system for obesogenic compounds: Screening of endocrine disrupting compounds and evaluation of the PPAR dependency of the effect. PLoS ONE 2013, 8, e77481. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Fernandes, A.; Vanparys, C.; Vergauwen, L.; Knapen, D.; Jorens, P.G.; Blust, R. Toxicogenomics in the 3T3-L1 cell line, a new approach for screening of obesogenic compounds. Toxicol. Sci. 2014, 140, 352–363. [Google Scholar] [CrossRef]
- Mangum, L.H.; Howell, G.E., 3rd; Chambers, J.E. Exposure to p,p’-DDE enhances differentiation of 3T3-L1 preadipocytes in a model of sub-optimal differentiation. Toxicol. Lett. 2015, 238, 65–71. [Google Scholar] [CrossRef]
- Peris-Sampedro, F.; Basaure, P.; Reverte, I.; Cabre, M.; Domingo, J.L.; Colomina, M.T. Chronic exposure to chlorpyrifos triggered body weight increase and memory impairment depending on human apoE polymorphisms in a targeted replacement mouse model. Physiol. Behav. 2015, 144, 37–45. [Google Scholar] [CrossRef]
- Traboulsi, H.; Guerrina, N.; Iu, M.; Maysinger, D.; Ariya, P.; Baglole, C.J. Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter. Int. J. Mol. Sci. 2017, 18, 243. [Google Scholar] [CrossRef]
- Bevan, G.H.; Al-Kindi, S.G.; Brook, R.; Rajagopalan, S. Ambient Air Pollution and Atherosclerosis: Recent Updates. Curr. Atheroscler. Rep. 2021, 23, 63. [Google Scholar] [CrossRef]
- Samet, J.M.; Dominici, F.; Curriero, F.C.; Coursac, I.; Zeger, S.L. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N. Engl. J. Med. 2000, 343, 1742–1749. [Google Scholar] [CrossRef]
- Vineis, P.; Husgafvel-Pursiainen, K. Air pollution and cancer: Biomarker studies in human populations. Carcinogenesis 2005, 26, 1846–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorntorp, P.; Rosmond, R. Obesity and cortisol. Nutrition 2000, 16, 924–936. [Google Scholar] [CrossRef]
- Harris, R.B. Chronic and acute effects of stress on energy balance: Are there appropriate animal models? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R250–R265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elobeid, M.A.; Padilla, M.A.; Brock, D.W.; Ruden, D.M.; Allison, D.B. Endocrine disruptors and obesity: An examination of selected persistent organic pollutants in the NHANES 1999-2002 data. Int. J. Environ. Res. Public Health 2010, 7, 2988–3005. [Google Scholar] [CrossRef] [Green Version]
- Turyk, M.E.; Anderson, H.A.; Steenport, D.; Buelow, C.; Imm, P.; Knobeloch, L. Longitudinal biomonitoring for polybrominated diphenyl ethers (PBDEs) in residents of the Great Lakes basin. Chemosphere 2010, 81, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Dirinck, E.; Jorens, P.G.; Covaci, A.; Geens, T.; Roosens, L.; Neels, H.; Mertens, I.; Van Gaal, L. Obesity and persistent organic pollutants: Possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity 2011, 19, 709–714. [Google Scholar] [CrossRef]
- Dhooge, W.; Den Hond, E.; Koppen, G.; Bruckers, L.; Nelen, V.; Van De Mieroop, E.; Bilau, M.; Croes, K.; Baeyens, W.; Schoeters, G.; et al. Internal exposure to pollutants and body size in Flemish adolescents and adults: Associations and dose-response relationships. Environ. Int. 2010, 36, 330–337. [Google Scholar] [CrossRef]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [Green Version]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Pesticide and insect repellent mixture (permethrin and DEET) induces epigenetic transgenerational inheritance of disease and sperm epimutations. Reprod. Toxicol. 2012, 34, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS ONE 2012, 7, e46249. [Google Scholar] [CrossRef]
- Tracey, R.; Manikkam, M.; Guerrero-Bosagna, C.; Skinner, M.K. Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reprod. Toxicol. 2013, 36, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K.; Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Haque, M.; Nilsson, E.E. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. 2013, 11, 228. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 2011, 6, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K.; Manikkam, M.; Haque, M.M.; Zhang, B.; Savenkova, M.I. Epigenetic transgenerational inheritance of somatic transcriptomes and epigenetic control regions. Genome Biol. 2012, 13, R91. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Lee, Y.A.; Hong, Y.C.; Cho, J.; Lee, K.S.; Shin, C.H.; Kim, B.N.; Kim, J.I.; Park, S.J.; Bisgaard, H.; et al. Effect of prenatal bisphenol A exposure on early childhood body mass index through epigenetic influence on the insulin-like growth factor 2 receptor (IGF2R) gene. Environ. Int. 2020, 143, 105929. [Google Scholar] [CrossRef]
- Manikkam, M.; Haque, M.M.; Guerrero-Bosagna, C.; Nilsson, E.E.; Skinner, M.K. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult-onset disease through the female germline. PLoS ONE 2014, 9, e102091. [Google Scholar] [CrossRef] [Green Version]
- Ozgyin, L.; Erdos, E.; Bojcsuk, D.; Balint, B.L. Nuclear receptors in transgenerational epigenetic inheritance. Prog. Biophys. Mol. Biol. 2015, 118, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J. Mol. Biol. 2014, 426, 3426–3441. [Google Scholar] [CrossRef]
- Doherty, L.F.; Bromer, J.G.; Zhou, Y.; Aldad, T.S.; Taylor, H.S. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm. Cancer 2010, 1, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Jacobs, D.R., Jr.; Porta, M. Hypothesis: A unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ. Health Perspect. 2009, 117, 1799–1802. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Endocrine Disruptor | Description | Obesogenic Actions | Epigenetic Effects | Refs. |
|---|---|---|---|---|
| Bisphenol A (BPA) | Synthetic organic compound used in polycarbonate and resins. Commonly detected in water bottles, food containers, and metal-based cans. | Stimulation of adipogenesis Induction of insulin resistance Alteration of pancreatic beta-cell function Hepatotoxicity Induction of adulthood hepatic steatosis Reduction in mitochondrial function | Reduction in global DNA methylation Changes in histone marks (H3Ac, H4Ac, H3K4me2, H3K36me3) | [60] [61] [62] |
| Diethylstilbestrol (DES) | Synthetic estrogen used to prevent adverse pregnancy outcomes. | Stimulation of markers of adiposity (leptin and proinflammatory cytokines, [IL-6]) Alteration in glucose metabolism and pancreatic beta-cell hyperplasia | Increased expression of long non-coding RNA HOTAIR. | [63] [64] [65] [66] |
| Phthalates | Diesters of phthalic acid, widely used in the production of plastic products (children’s toys, food packaging, medical devices, and furnishings). | Increased adipogenesis and insulin resistance Strong correlation between urinary levels of phthalates’ metabolites and obesity | Increased DNA methylation at level of genes related to metabolism Increased expression of miR-34a-5p and of long non-coding RNA H19 and its downstream pathway | [67] [68] [69] [67] [70] |
| Organochlorine (OCPs) and Organophosphate (OPPs) Pesticides | OCPs are chlorinated hydrocarbons used from the 1940s to the 1960s and are still detected in tap water. OPPs represent up to 50% of all the insecticide use worldwide. | Stimulation of adipogenesis OPPs accumulate in adipose tissue and influence PPARγ gene expression and production of inflammatory cytokines | Decreased global DNA methylation in both adipose-derived stromal cells (ADSCs) and 3T3-L1 preadipocytes Increased demethylation of lysine 27 on histone H3 (H3K27me3) | [71] [72] [73] [60] [74] [75] |
| Inhaled pollutants | Toxic environmental particles originate from a variety of sources (industrial pollution, automobile traffic, natural disasters). | Stimulation of the classic systemic inflammatory response associated with obesity, type 2 diabetes, insulin resistance, and metabolic syndrome | Altered DNA methylation status of PPARγ and PPARγ target genes | [76] [77] [78] [79] |
| Flame retardants | Group of compounds that prevent or slow the further development of ignition. | Stimulation of adipocytes differentiation Strong association between polybrominated diphenyl ethers (PBDEs) exposure and body mass index | Reduction in global DNA methylation | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nettore, I.C.; Franchini, F.; Palatucci, G.; Macchia, P.E.; Ungaro, P. Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Obesity. Biomedicines 2021, 9, 1716. https://doi.org/10.3390/biomedicines9111716
Nettore IC, Franchini F, Palatucci G, Macchia PE, Ungaro P. Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Obesity. Biomedicines. 2021; 9(11):1716. https://doi.org/10.3390/biomedicines9111716
Chicago/Turabian StyleNettore, Immacolata Cristina, Fabiana Franchini, Giuseppe Palatucci, Paolo Emidio Macchia, and Paola Ungaro. 2021. "Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Obesity" Biomedicines 9, no. 11: 1716. https://doi.org/10.3390/biomedicines9111716
APA StyleNettore, I. C., Franchini, F., Palatucci, G., Macchia, P. E., & Ungaro, P. (2021). Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Obesity. Biomedicines, 9(11), 1716. https://doi.org/10.3390/biomedicines9111716