
Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data

 , ,
, ,  ,
, 
Abstract
:1. Introduction
- Providing a thorough review about DL-based predictive models in epigenomics for disease detection, subtype classification, and treatment response prediction.
- Giving an insight into the main characteristics of the most common epigenomic data types and potential data sources, especially several publicly available databases, which could be used to develop the predictive models.
- Discussing data preprocessing flows, DL architectures, DL libraries, and model evaluation metrics that were feasible for epigenomics.
- Proposing current practical challenges and future trends of the development of epigenomic data-based DL techniques for translational medicine.
2. Materials and Methods
2.1. Search Strategy
2.2. Study Selection and Eligibility Criteria
- (1)
- DL models or predictive models that utilized DL as a component to solve human diseases-related tasks;
- (2)
- Prediction tasks directly targeted clinical outcomes of human diseases (i.e., disease detection, subtype classification, prognosis, and treatment response prediction). We excluded articles that addressed biological mechanisms of diseases such as genes and gene sets prediction, characterization of chromatin states, and miRNA-disease associations.
- (3)
- We focused on prediction applicability of epigenomic data including DNA methylation, histone modification, and non-coding RNA. Models using other omics data such as genomics, proteomics, transcriptomics, or multi-omics data were deleted;
- (4)
- Only original works were included. Reviews, commentaries, and editorials were excluded;
- (5)
- Publications with unavailable full texts were discarded.
2.3. Data Extraction
3. Results
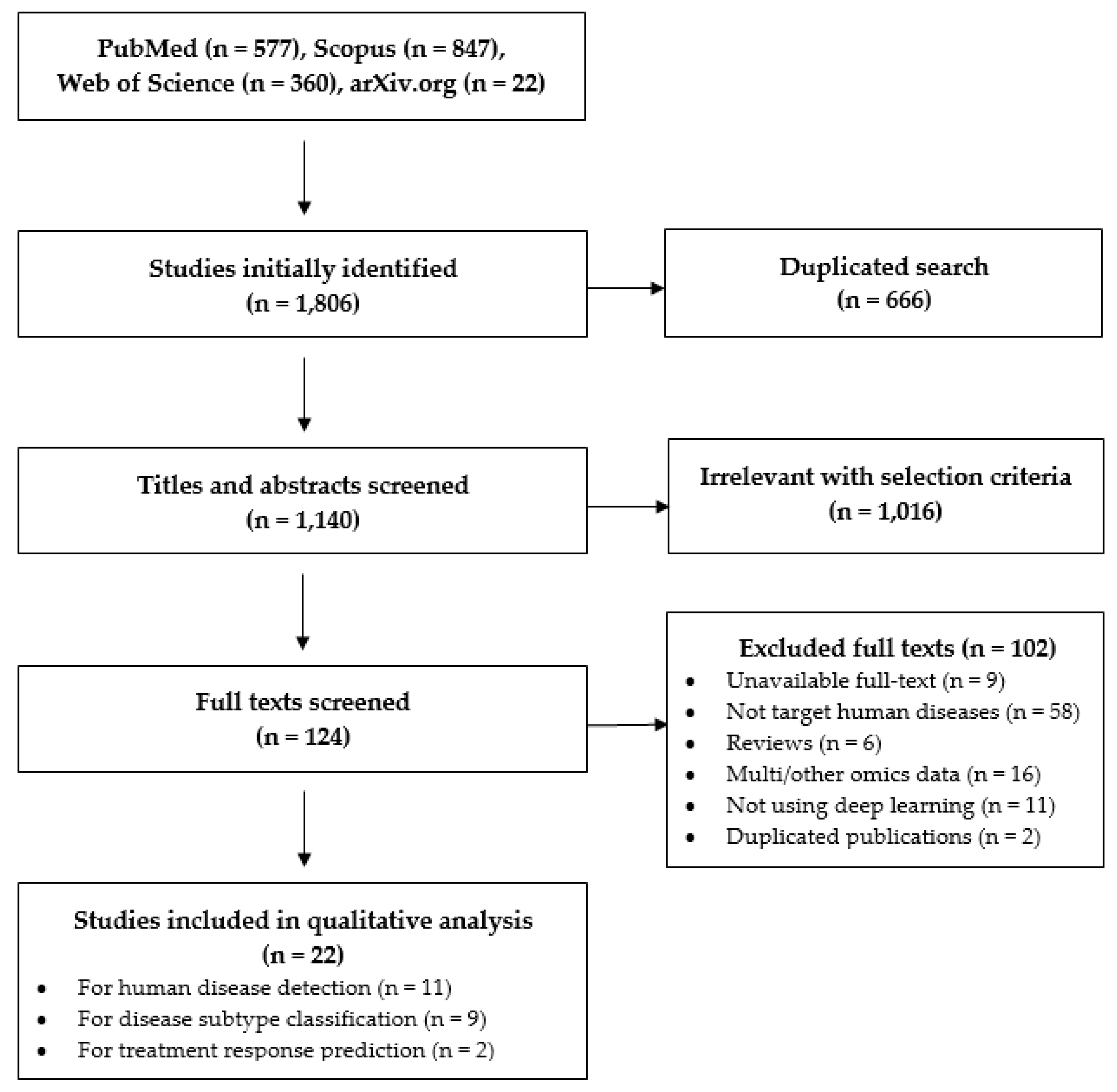
3.1. Selection Results
3.2. An Overview of DL in Translational Epigenomics
3.2.1. DL in Epigenomics for Disease Detection
- DNN-based extracted features were more effective for clustering analysis than those extracted from the principal component analysis (PCA) and non-negative matrix factorization (NMF),
- DERC achieved higher accuracy in breast cancer classification in comparison with other models under the same conditions.
3.2.2. DL in Epigenomics for Disease Subtype Classification
3.2.3. DL in Epigenomics for Treatment Response Prediction
3.3. An Insight into Epigenomic Data Used to Train Predictive Models for Human Diseases
3.3.1. Types of Epigenomic Data
3.3.2. Epigenomic Data Sources
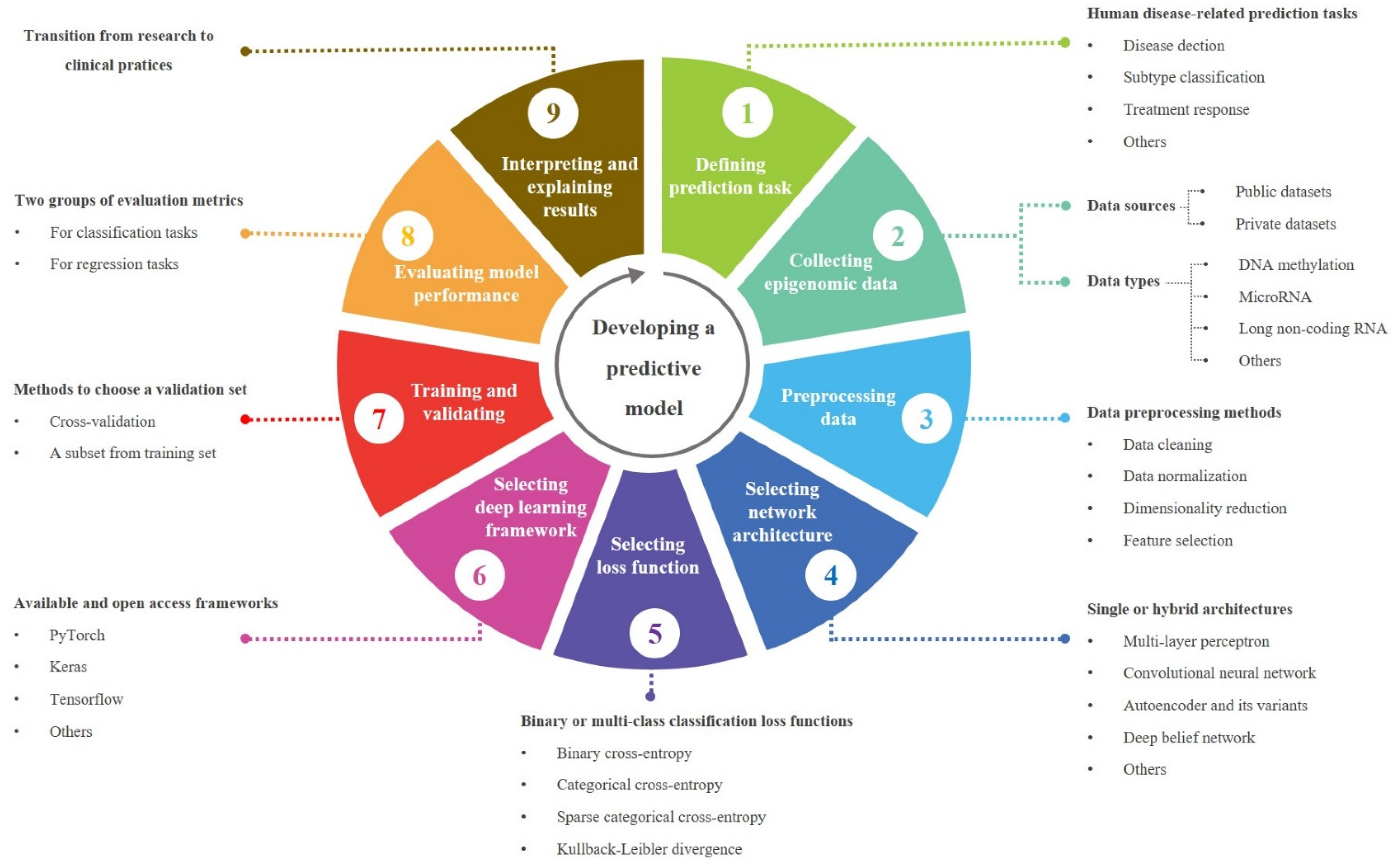
3.4. A Workflow for Developing a Predictive Model in Translational Epigenomics
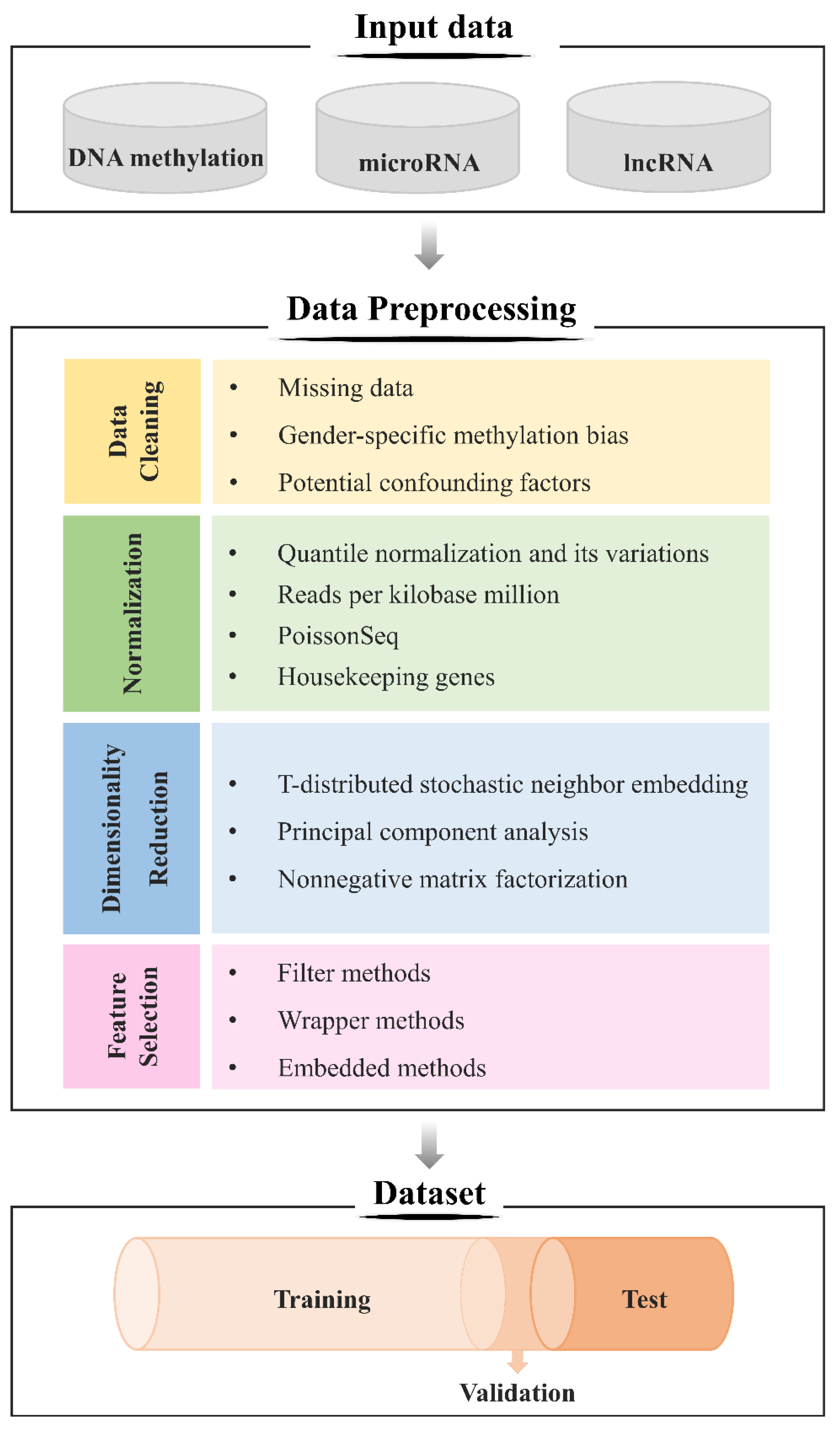
3.4.1. Data Preprocessing
3.4.2. Loss Function
3.4.3. Network Architectures
3.4.4. DL Libraries
3.4.5. Model Evaluation Metrics
4. Challenges and Future Research Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.C.; Papandreou, G.; Kokkinos, I.; Murphy, K.; Yuille, A.L. DeepLab: Semantic image segmentation with deep convolutional nets, atrous convolution, and fully connected CRFs. IEEE Trans. Pattern Anal. Mach. Intell. 2018, 40, 834–848. [Google Scholar] [CrossRef]
- Bengio, Y.; Courville, A.; Vincent, P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Y.; Liao, X.; Shi, W.; Li, K.; Zou, Q.; Peng, S. Deep learning in omics: A survey and guideline. Brief. Funct. Genom. 2019, 18, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Beaudet, A.L. Epigenetics and human disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a019497. [Google Scholar] [CrossRef]
- Tollefsbol, T.O. Chapter 1—An Overview of Epigenetics. In Handbook of Epigenetics, 2nd ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–6. [Google Scholar]
- Jung, M.; Pfeifer, G.P. CpG Islands. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Academic Press: San Diego, CA, USA, 2013; pp. 205–207. [Google Scholar]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as biomarkers in disease: Latest findings regarding their role in diagnosis and prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gimenez, J.L.; Seco-Cervera, M.; Tollefsbol, T.O.; Roma-Mateo, C.; Peiro-Chova, L.; Lapunzina, P.; Pallardo, F.V. Epigenetic biomarkers: Current strategies and future challenges for their use in the clinical laboratory. Crit. Rev. Clin. Lab. Sci. 2017, 54, 529–550. [Google Scholar] [CrossRef]
- Replogle, J.M.; De Jager, P.L. Epigenomics in translational research. Transl. Res. 2015, 165, 7–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukder, A.; Barham, C.; Li, X.; Hu, H. Interpretation of deep learning in genomics and epigenomics. Brief. Bioinform. 2020, 22, bbaa177. [Google Scholar] [CrossRef]
- Arslan, E.; Schulz, J.; Rai, K. Machine learning in epigenomics: Insights into cancer biology and medicine. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188588. [Google Scholar] [CrossRef] [PubMed]
- Brasil, S.; Neves, C.J.; Rijoff, T.; Falcao, M.; Valadao, G.; Videira, P.A.; Dos Reis Ferreira, V. Artificial intelligence in epigenetic studies: Shedding light on rare diseases. Front. Mol. Biosci. 2021, 8, 648012. [Google Scholar] [CrossRef]
- Rauschert, S.; Raubenheimer, K.; Melton, P.E.; Huang, R.C. Machine learning and clinical epigenetics: A review of challenges for diagnosis and classification. Clin. Epigenet. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holder, L.B.; Haque, M.M.; Skinner, M.K. Machine learning for epigenetics and future medical applications. Epigenetics 2017, 12, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Chen, Y.; Luo, C.; Meng, F. Machine learning methods in precision medicine targeting epigenetic diseases. Curr. Pharm. Des. 2018, 24, 3998–4006. [Google Scholar] [CrossRef] [PubMed]
- Iesato, A.; Nucera, C. Role of regulatory non-coding RNAs in aggressive thyroid cancer: Prospective applications of neural network analysis. Molecules 2021, 26, 3022. [Google Scholar] [CrossRef]
- Jovcevska, I. Next generation sequencing and machine learning technologies are painting the epigenetic portrait of glioblastoma. Front. Oncol. 2020, 10, 798. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Grp, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Bmj-Brit. Med. J. 2009, 339, b2535. [Google Scholar] [CrossRef] [Green Version]
- Afshar, S.; Afshar, S.; Warden, E.; Manochehri, H.; Saidijam, M. Application of artificial neural network in miRNA biomarker selection and precise diagnosis of colorectal cancer. Iran Biomed. J. 2019, 23, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh Savareh, B.; Asadzadeh Aghdaie, H.; Behmanesh, A.; Bashiri, A.; Sadeghi, A.; Zali, M.; Shams, R. A machine learning approach identified a diagnostic model for pancreatic cancer through using circulating microRNA signatures. Pancreatology 2020, 20, 1195–1204. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision cardiovascular medicine: Artificial intelligence and epigenetics for the pathogenesis and prediction of coarctation in neonates. J. Matern.-Fetal Neonatal Med. 2020, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Er, A.; Aydas, B.; Turkoglu, O.; Taskin, B.D.; Duman, M.; Yilmaz, D.; Radhakrishna, U. Artificial intelligence and the detection of pediatric concussion using epigenomic analysis. Brain Res. 2020, 1726, 146510. [Google Scholar] [CrossRef] [PubMed]
- del Amor, R.; Colomer, A.; Monteagudo, C.; Naranjo, N. A deep embedded refined clustering approach for breast cancer distinction based on DNA methylation. Neural Comput. Appl. 2021. [Google Scholar] [CrossRef]
- Duan, X.R.; Yang, Y.L.; Tan, S.J.; Wang, S.H.; Feng, X.L.; Cui, L.X.; Feng, F.F.; Yu, S.C.; Wang, W.; Wu, Y.J. Application of artificial neural network model combined with four biomarkers in auxiliary diagnosis of lung cancer. Med. Biol. Eng. Comput. 2017, 55, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Elias, K.M.; Fendler, W.; Stawiski, K.; Fiascone, S.J.; Vitonis, A.F.; Berkowitz, R.S.; Frendl, G.; Konstantinopoulos, P.; Crum, C.P.; Kedzierska, M.; et al. Diagnostic potential for a serum miRNA neural network for detection of ovarian cancer. Elife 2017, 6, e28932. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA methylation markers for pan-cancer prediction by deep learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Z.; Yu, H.; Ma, Z. Learning deep features for DNA methylation data analysis. IEEE Access 2016, 4, 2732–2737. [Google Scholar] [CrossRef]
- Xia, C.; Xiao, Y.; Wu, J.; Zhao, X.; Li, H. A convolutional neural network based ensemble method for cancer prediction using DNA methylation data. In Proceedings of the 2019 11th International Conference on Machine Learning and Computing, Zhuhai, China, 22–24 February 2019; pp. 191–196. [Google Scholar]
- Zhang, M.; Pan, C.; Liu, H.; Zhang, Q.; Li, H. An attention-based deep learning method for schizophrenia patients classification using DNA methylation data. In Proceedings of the 2020 42nd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Montreal, QC, Canada, 20–24 July 2020; pp. 172–175. [Google Scholar] [CrossRef]
- Al Mamun, A.; Mondal, A.M. Long non-coding RNA based cancer classification using deep neural networks. In Proceedings of the ACM-BCB 2019—10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; p. 541. [Google Scholar]
- Albaradei, S.; Thafar, M.; Van Neste, C.; Essack, M.; Bajic, V.B. Metastatic state of colorectal cancer can be accurately predicted with methylome. In Proceedings of the 2019 6th International Conference on Bioinformatics Research and Applications, Seoul, Korea, 19–21 December 2019; pp. 125–130. [Google Scholar]
- Batbaatar, E.; Park, K.H.; Amarbayasgalan, T.; Davagdorj, K.; Munkhdalai, L.; Pham, V.H.; Ryu, K.H. Class-incremental learning with deep generative feature replay for DNA methylation-based cancer classification. IEEE Access 2020, 8, 210800–210815. [Google Scholar] [CrossRef]
- Laplante, J.F.; Akhloufi, M.A. Predicting cancer types from miRNA stem-loops using deep learning. In Proceedings of the 2020 42nd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Montreal, QC, Canada, 20–24 July 2020; pp. 5312–5315. [Google Scholar] [CrossRef]
- Levy, J.J.; Titus, A.J.; Petersen, C.L.; Chen, Y.; Salas, L.A.; Christensen, B.C. MethylNet: An automated and modular deep learning approach for DNA methylation analysis. BMC Bioinform. 2020, 21, 108. [Google Scholar] [CrossRef] [Green Version]
- Smolander, J.; Stupnikov, A.; Glazko, G.; Dehmer, M.; Emmert-Streib, F. Comparing biological information contained in mRNA and non-coding RNAs for classification of lung cancer patients. BMC Cancer 2019, 19, 1176. [Google Scholar] [CrossRef] [Green Version]
- Titus, A.J.; Bobak, C.A.; Christensen, B.C. A new dimension of breast cancer epigenetics applications of variational autoencoders with DNA methylation. In Proceedings of the BIOINFORMATICS 2018—9th International Conference on Bioinformatics Models, Methods and Algorithms, Proceedings; Part of 11th International Joint Conference on Biomedical Engineering Systems and Technologies, BIOSTEC 2018, Funchal, Portugal, 19–21 January 2018; pp. 140–145. [Google Scholar]
- Wang, Z.; Wang, Y. Extracting a biologically latent space of lung cancer epigenetics with variational autoencoders. BMC Bioinform. 2019, 20, 568. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Xu, R. Predicting cancer origins with a DNA methylation-based deep neural network model. PLoS ONE 2020, 15, e0226461. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, L.H.-C.; Chen, B.-S. Investigating core signaling pathways of hepatitis b virus pathogenesis for biomarkers identification and drug discovery via systems biology and deep learning method. Biomedicines 2020, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Morilla, I.; Uzzan, M.; Laharie, D.; Cazals-Hatem, D.; Denost, Q.; Daniel, F.; Belleannee, G.; Bouhnik, Y.; Wainrib, G.; Panis, Y.; et al. Colonic microrna profiles, identified by a deep learning algorithm, that predict responses to therapy of patients with acute severe ulcerative colitis. Clin. Gastroenterol. Hepatol. 2019, 17, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Kan, X. Neural network cascade optimizes microRNA biomarker selection for nasopharyngeal cancer prognosis. PLoS ONE 2014, 9, e110537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Teschendorff, A.E.; Leijon, A.; Qiao, Y.; Zhang, H.; Guo, J. Variational bayesian matrix factorization for bounded support data. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 876–889. [Google Scholar] [CrossRef]
- Taghia, J.; Leijon, A. Variational inference for watson mixture model. IEEE Trans. Pattern Anal. Mach. Intell. 2016, 38, 1886–1900. [Google Scholar] [CrossRef]
- Ma, Z.; Leijon, A. Bayesian estimation of beta mixture models with variational inference. IEEE Trans. Pattern Anal. Mach. Intell. 2011, 33, 2160–2173. [Google Scholar] [CrossRef]
- Gentleman, R.; Carey, V.; Huber, W.; Irizarry, R.; Dudoit, S. Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Acurzio, B.; Verma, A.; Polito, A.; Giaccari, C.; Cecere, F.; Fioriniello, S.; Della Ragione, F.; Fico, A.; Cerrato, F.; Angelini, C.; et al. Zfp57 inactivation illustrates the role of ICR methylation in imprinted gene expression during neural differentiation of mouse ESCs. Sci. Rep. 2021, 11, 13802. [Google Scholar] [CrossRef]
- Paulsen, M.; Ferguson-Smith, A.C. DNA methylation in genomic imprinting, development, and disease. J. Pathol. 2001, 195, 97–110. [Google Scholar] [CrossRef]
- Siegmund, K.D. Statistical approaches for the analysis of DNA methylation microarray data. Hum. Genet. 2011, 129, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA regulation of epigenetic modifiers in breast cancer. Cancers 2019, 11, 897. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Gao, G.; Cao, Y. Long noncoding RNAs as novel biomarkers have a promising future in cancer diagnostics. Dis. Mark. 2016, 2016, 9085195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [Green Version]
- Armeev, G.A.; Kniazeva, A.S.; Komarova, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Histone dynamics mediate DNA unwrapping and sliding in nucleosomes. Nat. Commun. 2021, 12, 2387. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.; Li, M.; Yang, F.; Ma, Y.; Sang, J.; Li, R.; Li, Z.; Zhang, Z.; Bao, Y. EWAS Data Hub: A resource of DNA methylation array data and metadata. Nucleic Acids Res. 2020, 48, D890–D895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhao, N.; Zhang, X.; Xiao, J.; Li, J.; Lv, D.; Zhou, W.; Li, Y.; Xu, J.; Li, X. SurvivalMeth: A web server to investigate the effect of DNA methylation-related functional elements on prognosis. Brief. Bioinform. 2021, 22, bbaa162. [Google Scholar] [CrossRef] [PubMed]
- Grunau, C.; Renault, E.; Rosenthal, A.; Roizes, G. MethDB—A public database for DNA methylation data. Nucleic Acids Res. 2001, 29, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Rahm, E.; Do, H. Data Cleaning: Problems and current approaches. IEEE Data Eng. Bull. 2000, 23, 3–13. [Google Scholar]
- Wang, C.; Li, J. Deep learning framework identifies pathogenic noncoding somatic mutations from personal prostate cancer genomes. Cancer Res 2020, 80, 4644–4654. [Google Scholar] [CrossRef]
- Daca-Roszak, P.; Pfeifer, A.; Żebracka-Gala, J.; Rusinek, D.; Szybińska, A.; Jarząb, B.; Witt, M.; Ziętkiewicz, E. Impact of SNPs on methylation readouts by Illumina infinium humanMethylation450 beadchip array: Implications for comparative population studies. BMC Genom. 2015, 16, 1003. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Singh, B. Investigating the impact of data normalization on classification performance. Appl. Soft Comput. 2020, 97, 105524. [Google Scholar] [CrossRef]
- Wilhelm-Benartzi, C.S.; Koestler, D.C.; Karagas, M.R.; Flanagan, J.M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; Houseman, E.A.; Brown, R. Review of processing and analysis methods for DNA methylation array data. Br. J. Cancer 2013, 109, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wong, L.; Goh, W.W.B. How to do quantile normalization correctly for gene expression data analyses. Sci. Rep. 2020, 10, 15534. [Google Scholar] [CrossRef]
- Fortin, J.P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief. Bioinform. 2018, 19, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Boyd, V.L.; Moody, K.I.; Karger, A.E.; Livak, K.J.; Zon, G.; Burns, J.W. Methylation-dependent fragment separation: Direct detection of DNA methylation by capillary electrophoresis of PCR products from bisulfite-converted genomic DNA. Anal. Biochem. 2006, 354, 266–273. [Google Scholar] [CrossRef]
- Wang, G.; Kossenkov, A.V.; Ochs, M.F. LS-NMF: A modified non-negative matrix factorization algorithm utilizing uncertainty estimates. BMC Bioinform. 2006, 7, 175. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.J.; Titus, A.J.; Salas, L.A.; Christensen, B.C. Pymethylprocess-convenient high-throughput preprocessing workflow for DNA methylation data. Bioinformatics 2019, 35, 5379–5381. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Molinaro, A.M.; Simon, R.; Pfeiffer, R.M. Prediction error estimation: A comparison of resampling methods. Bioinformatics 2005, 21, 3301–3307. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Murphy, K.P. Machine Learning: A Probabilistic Perspective; MIT Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Hecht, N. Theory of the backpropagation neural network. In Proceedings of the International 1989 Joint Conference on Neural Networks, Washington, DC, USA, 18–22 June 1989; Volume 591, pp. 593–605. [Google Scholar]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef]
- Svozil, D.; Kvasnicka, V.; Pospichal, J. Introduction to multi-layer feed-forward neural networks. Chemom. Intell. Lab. Syst. 1997, 39, 43–62. [Google Scholar] [CrossRef]
- Kramer, M.A. Nonlinear principal component analysis using autoassociative neural networks. AIChE J. 1991, 37, 233–243. [Google Scholar] [CrossRef]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the dimensionality of data with neural networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Way, G.P.; Greene, C.S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. In Proceedings of the Pacific Symposium on Biocomputing 2018, Fairmont Orchid, HI, USA, 3–7 January 2018; pp. 80–91. [Google Scholar]
- Ian Goodfellow, Y.B.A.A.C. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- He, H.; Garcia, E.A. Learning from imbalanced data. IEEE Trans. Knowl. Data Eng. 2009, 21, 1263–1284. [Google Scholar] [CrossRef]
- Bomane, A.; Gonçalves, A.; Ballester, P.J. Paclitaxel response can be predicted with interpretable multi-variate classifiers exploiting DNA-methylation and miRNA data. Front. Genet. 2019, 10, 1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albaradei, S.; Napolitano, F.; Thafar, M.A.; Gojobori, T.; Essack, M.; Gao, X. MetaCancer: A deep learning-based pan-cancer metastasis prediction model developed using multi-omics data. Comput. Struct. Biotechnol. J. 2021, 19, 4404–4411. [Google Scholar] [CrossRef]
- Park, C.; Ha, J.; Park, S. Prediction of Alzheimer’s disease based on deep neural network by integrating gene expression and DNA methylation dataset. Expert Syst. Appl. 2020, 140, 112873. [Google Scholar] [CrossRef]
- Hira, M.T.; Razzaque, M.A.; Angione, C.; Scrivens, J.; Sawan, S.; Sarker, M. Integrated multi-omics analysis of ovarian cancer using variational autoencoders. Sci. Rep. 2021, 11, 6265. [Google Scholar] [CrossRef]
- Baek, B.; Lee, H. Prediction of survival and recurrence in patients with pancreatic cancer by integrating multi-omics data. Sci. Rep. 2020, 10, 18951. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep inside convolutional networks: Visualising image classification models and saliency maps. arXiv 2013, arXiv:1312.6034. [Google Scholar]
- Bergstra, J.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for hyper-parameter optimization. In Proceedings of the 24th International Conference on Neural Information Processing Systems, Granada, Spain, 12–15 December 2011; pp. 2546–2554. [Google Scholar]
- Wang, H.; Pujos-Guillot, E.; Comte, B.; de Miranda, J.L.; Spiwok, V.; Chorbev, I.; Castiglione, F.; Tieri, P.; Watterson, S.; McAllister, R.; et al. Deep learning in systems medicine. Brief. Bioinform. 2021, 22, 1543–1559. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research | Title | Main Findings |
|---|---|---|
| Zhang et al. (2019) [3] | DL in Omics: A Survey and Guideline |
|
| Min et al. (2017) [4] | DL in Bioinformatics |
|
| Talukder et al. (2020) [12] | Interpretation of DL in Genomics and Epigenomics |
|
| Arslan et al. (2021) [13] | ML in Epigenomics: Insights into Cancer Biology and Medicine |
|
| Brasil et al. (2021) [14] | Artificial Intelligence in Epigenetic Studies: Shedding Light on Rare Diseases |
|
| Rauschert et al. (2020) [15] | ML and Clinical Eepigenetics: A Review of Challenges for Diagnosis and Classification |
|
| Holder et al. (2017) [16] | ML for Epigenetics and Future Medical Applications |
|
| Fan et al. (2018) [17] | ML Methods in Precision Medicine Targeting Epigenetic Diseases |
|
| Iestao et al. (2021) [18] | Role of Regulatory Non-Coding RNAs in Aggressive Thyroid Cancer: Prospective Applications of Neural Network Analysis |
|
| Jovčevska et al.(2020) [19] | Next Generation Sequencing and ML Technologies Are Painting the Epigenetic Portrait of Glioblastoma |
|
| Research | Country | Target Disease | Data Type | Epigenomic Data Source | Validation Scheme | Predictive Model | Evaluation Metrics | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Sensi-tivity | Speci-ficity | Accu-Racy | Preci-Sion | F1-Score | |||||||
| Afshar et al. (2019) [21] | Iran | Colorectal cancer | miRNA | 50 CS and 150 NS (GSE59856–GEO) | 15% of the dataset | ANN | 1.000 | 0.900 | 0.970 | 1.000 | – | – |
| Alizadeh et al. (2020) [22] | Iran | Pancreatic cancer | miRNA | GSE113486; GSE59856; GSE85589; GSE106817; GSE112264; GSE124158 (GEO) | 5-fold CV on training and testing sets | ANN + PSO | – | 0.930 | 0.920 | 0.930 | – | – |
| Amor et al. (2021) [25] | Spain | Breast cancer | DNAm | GSE32393; GSE57285; GSE50220 (GEO) | 10% of the dataset | VAE | – | – | – | 0.993 | – | – |
| Bahado-Singh et al. (2020) [23] | USA | Coarctation of the aorta | DNAm | 24 cases and 16 controls | 10-fold CV on training set (80% of the dataset) | DNN | 0.970 | 0.950 | 0.980 | – | – | – |
| Bahado-Singh et al. (2020) [24] | USA | Concussion | DNAm | 17 cases and 18 controls | 10-fold CV on training set (80% of the dataset) | DNN | 0.989 | 0.950 | 0.912 | – | – | – |
| Duan et al. (2017) [26] | China | Lung cancer | DNAm | 200 CS and 200 NS | – | Back-propagation NN | 0.760 | – | – | – | – | – |
| Elias et al. (2017) [27] | USA | Ovarian cancer | miRNA | 179 human serum samples | 51 independent clinical samples | MLP | 0.900 | – | 1.000 | – | 0.913 | – |
| Liu et al. (2019) [28] | China | Pan-cancer (27 types) | DNAm (CpG markers) | 10,140 CS and 3386 NS (TCGA and GEO) | 370/4840 CS, 201/1742 NS | Two multi-layer feedforward NNs | 0.989 | 0.928 | 0.901 | 0.924 | – | – |
| DNAm (Promoter markers) | 0.985 | 0.898 | 0.811 | 0.883 | – | – | ||||||
| Si et al. (2016) [29] | China | Breast cancer | DNAm | 113 CS and 23 NS (GSE32393–GEO) | – | Auto-encode DNN + SOM | – | – | – | 0.971 | – | – |
| Xia et al. (2019) [30] | China | LUAD | DNAm | 460 CS and 32 NS (TCGA) | 5-fold CV on the whole datasets | CNN based ensemble model | 0.998 | – | – | 0.994 | – | – |
| LIHC | 379 CS and 50 NS (TCGA) | 0.994 | – | – | 0.988 | – | – | |||||
| KIRC | 320 CS and 160 NS (TCGA) | 0.999 | – | – | 0.996 | – | – | |||||
| Zhang et al. (2020) [31] | China | Schizophrenia | DNAm | 54 cases and 18 controls | 10-fold CV on the whole dataset | Attention-based FC + DAE + SVM | – | 0.998 | 0.988 | 0.991 | – | – |
| Research | Country | Target Disease | Data Type | Epigenomic Data Source | Validation Scheme | Predictive Model | Evaluation Metrics | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Sensi-Tivity | Speci-Ficity | ACCU-RACY | Preci-Sion | F1-Score | |||||||
| Al Mamun et al. (2019) [32] | USA | 8 types of cancer | long ncRNA | UCSC xena (TCGA) | – | MLP | – | 0.929 | – | 0.937 | 0.932 | 0.939 |
| LSTM | – | 0.952 | – | 0.956 | 0.951 | 0.951 | ||||||
| CNN | – | 0.976 | – | 0.978 | 0.977 | 0.976 | ||||||
| DAE | – | 0.959 | – | 0.964 | 0.961 | 0.960 | ||||||
| Albaradei et al. (2019) [33] | Kingdom of Saudi Arabia | Colorectal cancer | DNAm | 300 samples (TCGA) | 15% of the dataset | CNN | – | 0.967 | 0.958 | 0.962 | 0.904 | 0.947 |
| Batbaatar et al. (2020) [34] | South Korea | 12 types of cancer | DNAm | 2728 samples (TCGA) | – | An incremental feature selection + a scholar network | – | – | – | 0.932 | – | – |
| Laplante et al. (2020) [35] | Canada | 27 types of cancer | miRNA stem-loops | 8573 cases (TCGA) | 15% of the dataset | ANN | – | 0.969 | – | 0.969 | 0.969 | 0.969 |
| Levy et al. (2020) [36] | USA | 32 types of cancer | DNAm | 8891 samples (TCGA) | 20% of the dataset | VAE + MLP | – | 0.970 | – | 0.970 | 0.970 | 0.970 |
| Smolander et al. (2019) [37] | Finland | Lung cancer | ncRNA | 62 cases and 62 controls (GSE40419–GEO) | 10-fold CV on the whole dataset | DBN | 0.968 | 1.00 | 0.936 | 0.968 | – | – |
| Titus et al. (2018) [38] | USA | Breast cancer | DNAm | 86 normal-adjacent samples (TCGA) | Training/validation = 90/10 | VAE + Logistic regression classifiers | – | – | – | 0.961 | – | – |
| 86 basal-like samples (TCGA) | – | – | – | 0.944 | – | – | ||||||
| 31 Her2 samples (TCGA) | – | – | – | 0.961 | – | – | ||||||
| 285 Luminal A samples (TCGA) | – | – | – | 0.695 | – | – | ||||||
| 124 Luminal B samples (TCGA) | – | – | – | 0.843 | – | – | ||||||
| Wang et al. (2019) [39] | China | Lung cancer | DNAm | 507 LUAD samples (TCGA) | Training/validation = 90/10 | VAE + Logistic regression classifiers | – | 0.990 | – | – | 0.920 | 0.960 |
| 412 LUSC samples (TCGA) | – | 0.960 | – | – | 0.990 | 0.970 | ||||||
| Zheng et al. (2020) [40] | USA | 18 types of cancer | DNAm | 7339 samples (TCGA) | 10-fold CV on training set (60% of the dataset) | MLP | – | 0.926 | 0.997 | – | 0.950 | – |
| Research | Country | Target Disease/Drug | Data Type | Epigenomic Data Source | Validation Scheme | DL Model | Evaluation Metrics | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Sensi-Tivity | Speci-Ficity | ACCU-RACY | Preci-Sion | F1-Score | |||||||
| Chang et al. (2020) [41] | Taiwan | Hepatitis B | RNA-seq | GSE101575 (GEO) | 10-fold CV on the whole dataset | FC neural network | 0.990 | – | – | 0.926 | – | – |
| Morilla et al. (2018) [42] | France | Ulcerative colitis | miRNA | 47 samples | Leave-one-out and K-fold CV on 29 samples | DNN | – | – | – | – | – | – |
| Steroids | 0.910 | – | – | 0.930 | – | – | ||||||
| Infliximab | 0.820 | – | – | 0.840 | – | – | ||||||
| Cyclosporine | 0.790 | – | – | 0.800 | – | – | ||||||
| Characteristics | Common Public Databases | Private Dataset | |
|---|---|---|---|
| The Cancer Genome Atlas | Gene Expression Omnibus | ||
| Target disease | Cancer only | Various diseases | Various diseases |
| Data type | Clinical, gene copy number, DNA, imaging, methylation, microsatellite instability, microRNA, messenger RNA expression, protein expression | Gene expression, non-coding RNA, chromatin immunoprecipitation, DNA methylation, real-time PCR, genome variation profiling, single nucleotide polymorphisms arrays, serial analysis of gene expression, protein array | Depends on study objective |
| Data format | Raw, normalized, integrated data | Both raw and processed data submitted by the researchers | Raw data |
| Data source | Specific studies | Samples collected directly from patients | |
| Library | Brief Description | Creator | Programming Language | Operating System | Links (accessed on 27 October 2021) |
|---|---|---|---|---|---|
| Deeplearning4J | Supports all the needs of the based DL application | Skymind | Python, Java, Scala, C++, C, CUDA | Linux, Win, OSX, Android | deeplearning4j.org |
| Keras | Focuses on enabling fast experimentation | Franois Chollet | Python, R, CUDA | Linux, Win, OSX | keras.io cran.r-project.org/web/packages/keras/ |
| H2O | The scalable open-source machine learning platform that offers parallelized implementation of many supervised and unsupervised learning algorithms | Erin LeDell et al. | R, Java | Win, OSX, Ubuntu | cran.r-project.org/web/ packages/h2o |
| PyTorch | An optimized tensor library for DL using graphics processing units and central processing units | Python, CUDA, C++ | Linux, Win, OSX | pytorch.org | |
| TensorFlow | Has a comprehensive and flexible suite of tools | Python, C++, GO, Java, R, CUDA | Linux, Win, OSX, Android | tensorflow.org tensorflow.rstudio.com | |
| Scikit-learn | Provides many supervised and unsupervised learning algorithms via a consistent interface | David Cournapeau et al. | Python, C, C++, Cython | Linux, Win, OSX | scikit-learn.org |
| Evaluation Metric | Definition | Strength | Limitation |
|---|---|---|---|
| Accuracy | Fraction of correctly classified instances in the test set. A complement to the error-rate that measures fraction of the instances from the test set that are misclassified by the learning algorithm. | Summarize the overall performance. | Not relevant when either the performance on different classes is of varying importance or the distribution of instances in the different classes of the test data is skewed. |
| True positive rate (Sensitivity or recall) | Proportion of actual positives which are correctly identified. | Ameliorates the effect of class imbalance arising in the accuracy or error-rate measurements thereby skewing these estimates. | In the case of a multiclass classification problem, this would lead to as many metrics as there are classes, making it difficult to interpret. |
| True negative rate (Specificity) | Proportion of actual negatives which are correctly identified. | ||
| Positive predictive value (Precision) | Proportion of relevant examples (true positives) among all of the examples which were predicted to belong in a certain class. | Gives an insight into how reliable the class-wise predictions of a classifier is. | Might not provide enough information for a concrete judgment call on the superiority of the classifier in one case or the other. |
| F-score | An even weighted harmonic mean of precision and recall. The most commonly used metric is F1-score that weights the recall and precision of the classifier evenly. | Leaves out the true-negative performance of the classifier. | Ignores true negatives and thus is misleading for unbalanced classes. Appropriate weights for combining the precision and recall are generally not known. |
| Receiver operator characteristics (ROC) curve | A plot which takes true positive rate as the vertical axis and false positive rate as the horizontal axis. | Visualizes the performance of classifiers over their operating ranges. | Unable to quantify the comparative analysis that can facilitate decision making with regard to the suitability or preference of one classifier over others in the form of an objective scalar metric. |
| Area under the ROC curve (AUC) | Entire two dimensional area underneath the entire ROC curve. | Provides an aggregate measure of performance across all possible classification thresholds. | Loses significant information about the behavior of the learning algorithm over the entire operating range. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.M.; Kim, N.; Kim, D.H.; Le, H.L.; Piran, M.J.; Um, S.-J.; Kim, J.H. Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data. Biomedicines 2021, 9, 1733. https://doi.org/10.3390/biomedicines9111733
Nguyen TM, Kim N, Kim DH, Le HL, Piran MJ, Um S-J, Kim JH. Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data. Biomedicines. 2021; 9(11):1733. https://doi.org/10.3390/biomedicines9111733
Chicago/Turabian StyleNguyen, Thi Mai, Nackhyoung Kim, Da Hae Kim, Hoang Long Le, Md Jalil Piran, Soo-Jong Um, and Jin Hee Kim. 2021. "Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data" Biomedicines 9, no. 11: 1733. https://doi.org/10.3390/biomedicines9111733
APA StyleNguyen, T. M., Kim, N., Kim, D. H., Le, H. L., Piran, M. J., Um, S. -J., & Kim, J. H. (2021). Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data. Biomedicines, 9(11), 1733. https://doi.org/10.3390/biomedicines9111733