
Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Analysis
2.2. Molecular Phylogenetic Analysis
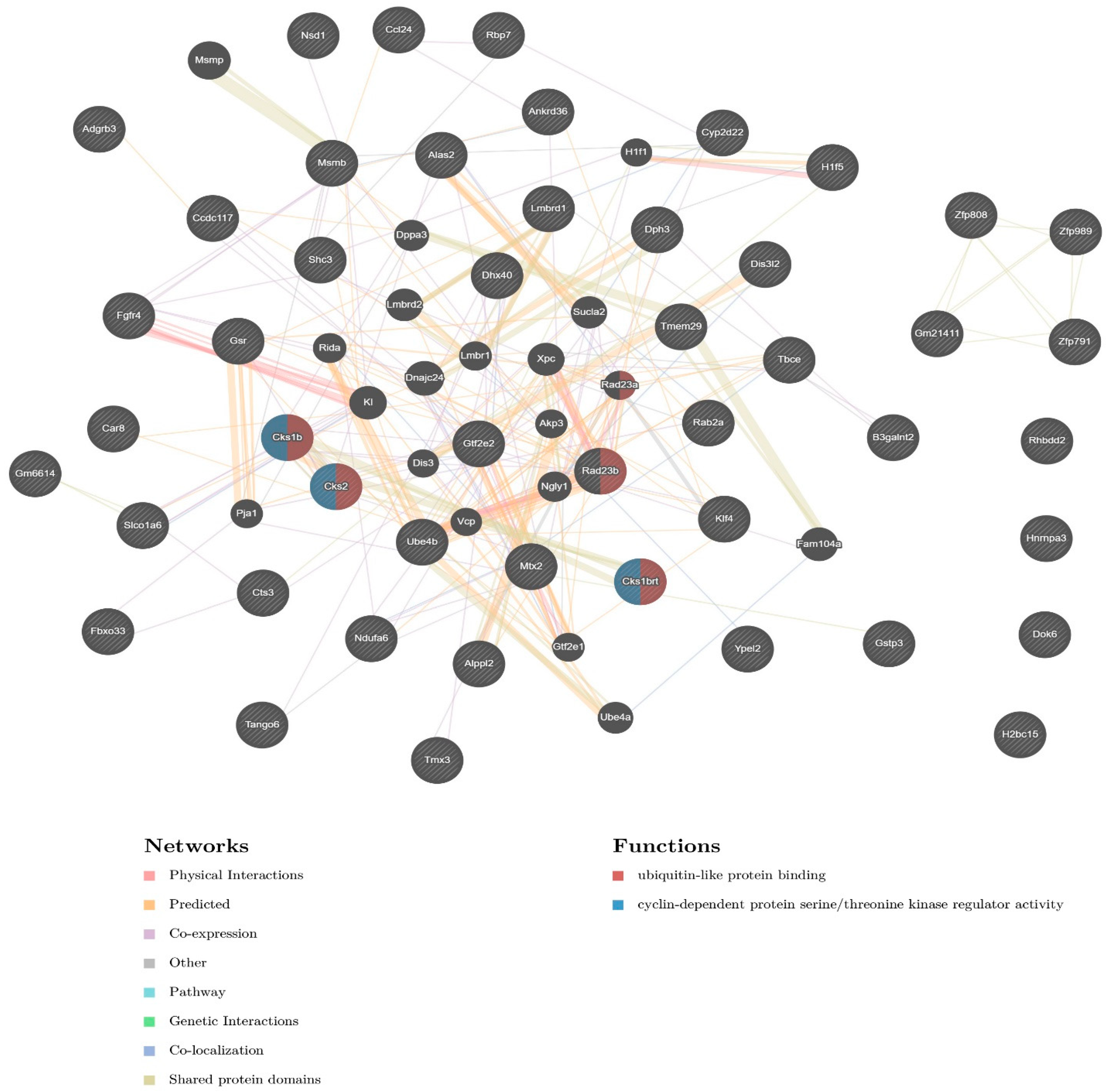
2.3. Pathway and Network Analysis
3. Results
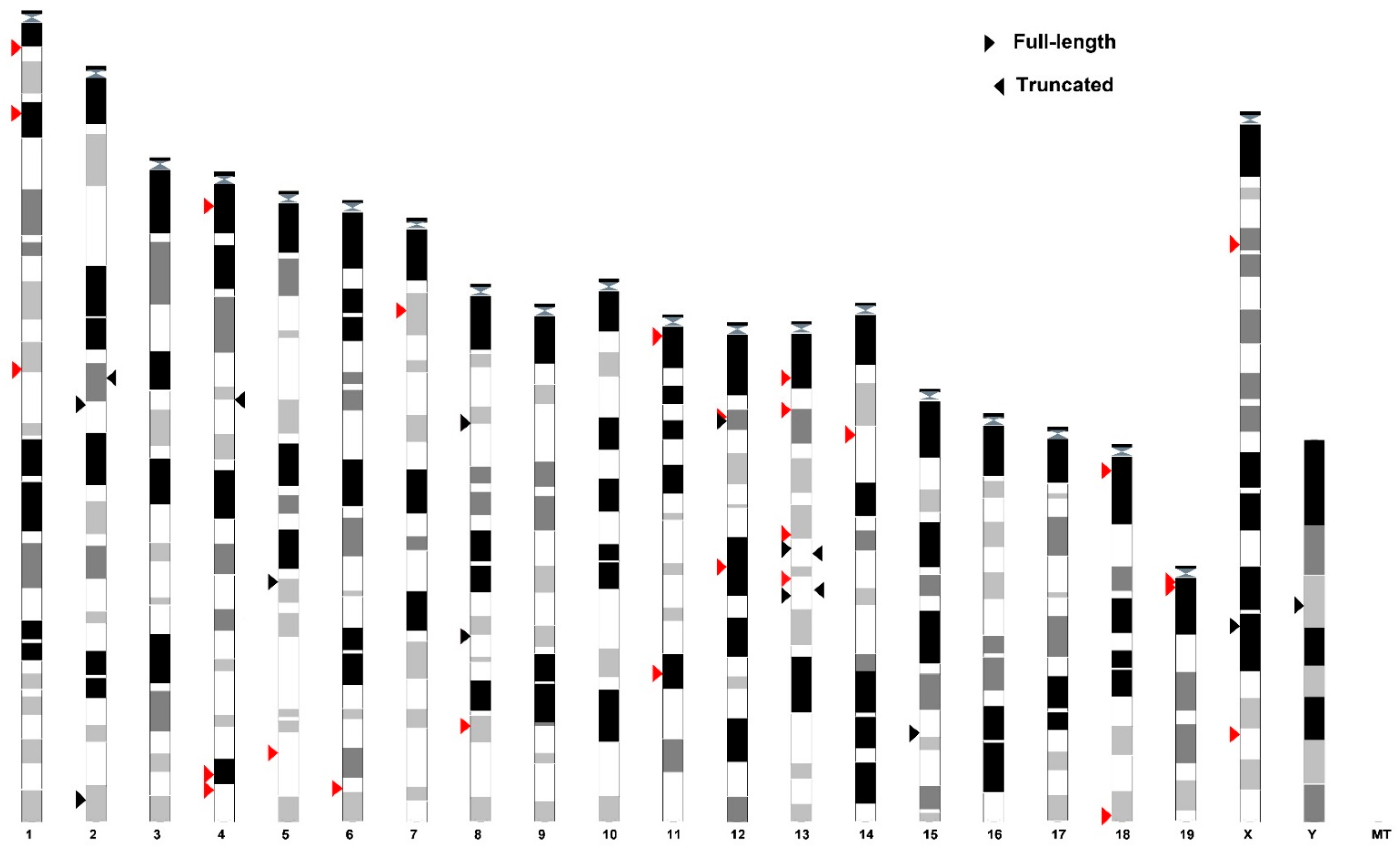
3.1. SEV Enrichment Motif Is a Universal Feature of VL30 Family
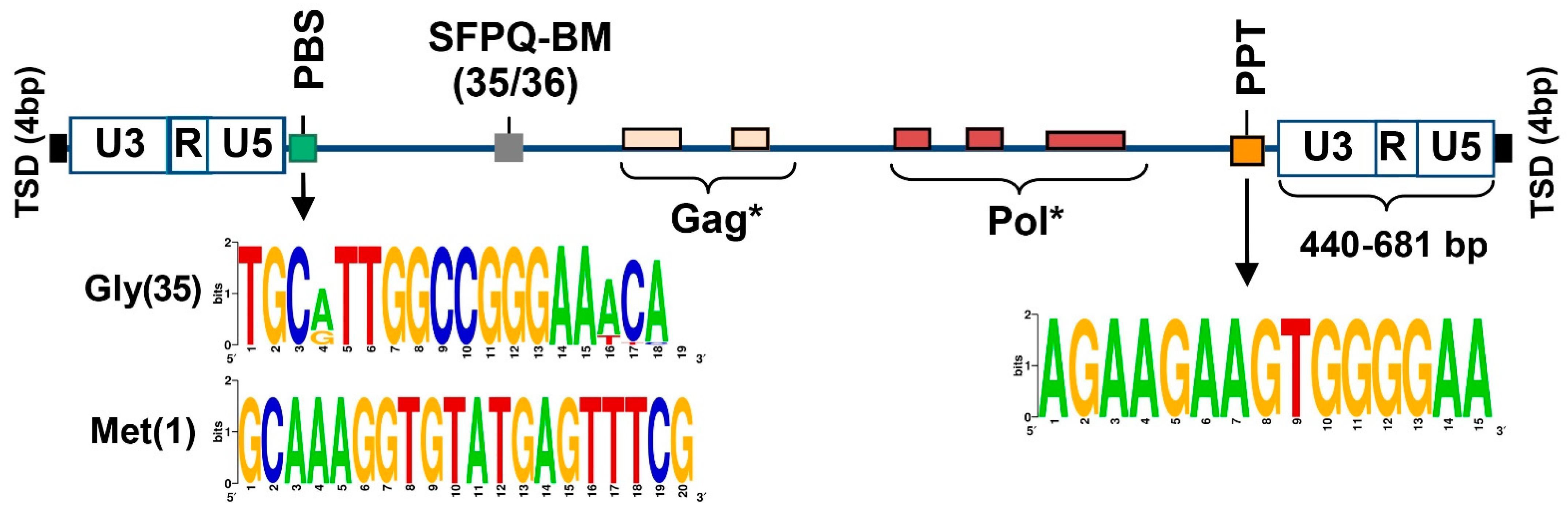
3.2. Structural and Functional Properties of SEV-VL30 RNAs
3.3. Insights into the Evolution of SEV-VL30s
3.4. Epigenetic Regulation of SEV-VL30s
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Zhu, G.; Xu, J.; Lai, Q.; Yan, B.; Guo, Y.; Fung, T.K.; Zeisig, B.B.; Cui, Y.; Zha, J.; et al. HOTTIP lncRNA Promotes Hematopoietic Stem Cell Self-Renewal Leading to AML-like Disease in Mice. Cancer Cell 2019, 36, 645–659.e8. [Google Scholar] [CrossRef]
- Lee, J.T.; Bartolomei, M.S. X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Qian, W.; Wang, S.; Ji, D.; Wang, Q.; Li, J.; Peng, W.; Gu, J.; Hu, T.; Ji, B.; et al. Analysis of lncRNA-Associated ceRNA Network Reveals Potential lncRNA Biomarkers in Human Colon Adenocarcinoma. Cell. Physiol. Biochem. 2018, 49, 1778–1791. [Google Scholar] [CrossRef]
- Song, J.; Ye, A.; Jiang, E.; Yin, X.; Chen, Z.; Bai, G.; Zhou, Y.; Liu, J. Reconstruction and analysis of the aberrant lncRNA-miRNA-mRNA network based on competitive endogenous RNA in CESC. J. Cell. Biochem. 2018, 119, 6665–6673. [Google Scholar] [CrossRef] [Green Version]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing miRNA–lncRNA interactions. In Long Non-Coding RNAs; Springer: Berlin/Heidelberg, Germany, 2016; pp. 271–286. [Google Scholar]
- Keshet, E.; Shaul, Y.; Kaminchik, J.; Aviv, H. Heterogeneity of “virus-like” genes encoding retrovirus-associated 30S RNA and their organization within the mouse genome. Cell 1980, 20, 431–439. [Google Scholar] [CrossRef]
- Markopoulos, G.; Noutsopoulos, D.; Mantziou, S.; Gerogiannis, D.; Thrasyvoulou, S.; Vartholomatos, G.; Kolettas, E.; Tzavaras, T. Genomic analysis of mouse VL30 retrotransposons. Mob. DNA 2016, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Konisti, S.; Mantziou, S.; Markopoulos, G.; Thrasyvoulou, S.; Vartholomatos, G.; Sainis, I.; Kolettas, E.; Noutsopoulos, D.; Tzavaras, T. H2O2 signals via iron induction of VL30 retrotransposition correlated with cytotoxicity. Free. Radic. Biol. Med. 2012, 52, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Noutsopoulos, D.; Markopoulos, G.; Vartholomatos, G.; Kolettas, E.; Kolaitis, N.; Tzavaras, T. VL30 retrotransposition signals activation of a caspase-independent and p53-dependent death pathway associated with mitochondrial and lysosomal damage. Cell Res. 2010, 20, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Tzavaras, T.; Kalogera, C.; Eftaxia, S.; Saragosti, S.; Pagoulatos, G.N. Clone-specific high-frequency retrotransposition of a recombinant virus containing a VL30 promoter in SV40-transformed NIH3T3 cells. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 1998, 1442, 186–198. [Google Scholar] [CrossRef]
- French, N.S.; Norton, J.D. Structure and functional properties of mouse VL30 retrotransposons. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 1997, 1352, 33–47. [Google Scholar] [CrossRef]
- Noutsopoulos, D.; Markopoulos, G.; Koliou, M.; Dova, L.; Vartholomatos, G.; Kolettas, E.; Tzavaras, T. Vanadium Induces VL30 Retrotransposition at an Unusually High Level: A Possible Carcinogenesis Mechanism. J. Mol. Biol. 2007, 374, 80–90. [Google Scholar] [CrossRef]
- Tzavaras, T.; Eftaxia, S.; Tavoulari, S.; Hatzi, P.; Angelidis, C. Factors influencing the expression of endogenous reverse transcriptases and viral-like 30 elements in mouse NIH3T3 cells. Int. J. Oncol. 2003, 23, 1237–1243. [Google Scholar] [CrossRef]
- Noutsopoulos, D.; Vartholomatos, G.; Kolaitis, N.; Tzavaras, T. SV40 Large T Antigen Up-regulates the Retrotransposition Frequency of Viral-like 30 Elements. J. Mol. Biol. 2006, 361, 450–461. [Google Scholar] [CrossRef]
- Thrasyvoulou, S.; Vartholomatos, G.; Markopoulos, G.; Noutsopoulos, D.; Mantziou, S.; Gkartziou, F.; Papageorgis, P.; Charchanti, A.; Kouklis, P.; Constantinou, A.I.; et al. VL30 retrotransposition is associated with induced EMT, CSC generation and tumorigenesis in HC11 mouse mammary stem-like epithelial cells. Oncol. Rep. 2020, 44, 126–138. [Google Scholar] [CrossRef]
- Song, X.; Sui, A.; Garen, A. Binding of mouse VL30 retrotransposon RNA to PSF protein induces genes repressed by PSF: Effects on steroidogenesis and oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Garen, A. From a retrovirus infection of mice to a long noncoding RNA that induces proto-oncogene transcription and oncogenesis via an epigenetic transcription switch. Signal Transduct. Target. Ther. 2016, 1, 16007. [Google Scholar] [CrossRef] [Green Version]
- Negahdaripour, M.; Owji, H.; Eskandari, S.; Zamani, M.; Vakili, B.; Nezafat, N. Small extracellular vesicles (sEVs): Discovery, functions, applications, detection methods and various engineered forms. Expert Opin. Biol. Ther. 2021, 21, 371–394. [Google Scholar] [CrossRef]
- Barrios, M.H.; Garnham, A.L.; Foers, A.D.; Cheng-Sim, L.; Masters, S.L.; Pang, K.C. Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation? Biomedicines 2021, 9, 1136. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [PubMed] [Green Version]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Kent, W.J. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.T.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser database: 2022 update. Nucleic Acids Res. 2021, 49, 1046. [Google Scholar] [CrossRef]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, 493D–496D. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Flicek, P. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Hubbard, T.; Barker, D.; Birney, E.; Cameron, G.; Chen, Y.; Clark, L.; Cox, T.; Cuff, J.; Curwen, V.; Down, T.; et al. The Ensembl genome database project. Nucleic Acids Res. 2002, 30, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- McLean, C.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q.D. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9, S4. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.J.; Jeon, M.; Stein, D.J.; Moiseyev, N.; Kropiwnicki, E.; Dai, C.; Xie, Z.; Wojciechowicz, M.L.; Litz, S.; Hom, J.; et al. Appyters: Turning Jupyter Notebooks into data-driven web apps. Gene Expr. Patterns 2021, 2, 100213. [Google Scholar] [CrossRef]
- Song, X.; Sun, Y.; Garen, A. From The Cover: Roles of PSF protein and VL30 RNA in reversible gene regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 12189–12193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, G.; Kimura, Y.; Daub, C.; Wani, S.; Plessy, C.; Irvine, K.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Ron, G.; Globerson, Y.; Moran, D.; Kaplan, T. Promoter-enhancer interactions identified from Hi-C data using probabilistic models and hierarchical topological domains. Nat. Commun. 2017, 8, 2237. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, T.; Preissl, S.; Amaral, M.L.; Grinstein, J.D.; Farah, E.N.; Destici, E.; Qiu, Y.; Hu, R.; Lee, A.Y.; et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019, 51, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L.; Kazazian, H.H., Jr. Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell 2008, 135, 23–35. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRE-VL30 | Nearest Genes (Distance to TSS) | cCRE Type/Function |
|---|---|---|
| 1qA5 | Lmbrd1 (+338,654), Adgrb3 (+812,423) | enhD (2X) |
| 1qD | Alppl2 (+11,819), Dis3l2 (+374,273) | enhD, CTCF |
| 2qC3tr | Hnrnpa3 (−18,737), Mtx2 (+814,721) | enhP (4X), enhD (5X) |
| 4qA1 | Car8 (−156,331), Rab2a (−140,272) | enhD |
| 4qB3tr | Rad23b (+70,666), Klf4 (+111,757) | CTCF |
| 4qE1-2 | Zfp989 (−32,805), Gm21,411 (−29,581) | K4m3, enhP |
| 4qE2 | Ube4b (−9840), Rbp7 (+18,389) | Prom, enhP (5X), enhD (6X) |
| 5qG2 | Ccl24 (−53,256), Rhbdd2 (−6313) | Prom, enhP (2X), CTCF (2X), K4m3 |
| 6qG2 | Gm6614 (−158,676), Slco1a6 (+16,063) | enhP, enhD |
| 8qA4 | Gtf2e2 (−16,097), Gsr (+63,004) | enhD (3X) |
| 8qC3 | Zfp791 (−16,620), Cks1brt (−12,209) | enhD (2X) |
| 8qD3 | Tango6 (−4073) | prom, EnhP, enhD (2X), CTCF (2X) |
| 11qA1 | Alas2 (−59,876), Tmem29 (−28,349) | enhP (4X), enhD (3X) |
| 11qC | Ankrd36 (−14,253), Ccdc117 (−13,244) | prom, enhP (2), enhD (4X) |
| 12qC1 | Dhx40 (−6323), Ypel2 (+179,638) | Prom, enhP (5X), enhD (2X) |
| 13qA1 | Fbxo33 (−5156) | enhD (6X) |
| 13qA3-1 | Tbce (+16,998), B3galnt2 (+68,171) | prom (3X), enhP (2X), enhD (2X) |
| 13qA5 | Hist1h2bn (+11,370), Hist1h1b (+15,132) | prom, enhP (5X), enhD, K4m3 |
| 13qB1 | Shc3 (−69,417), Cks2 (−8731) | enhD (7X), CTCF |
| 13qB3tr | Nsd1 (−22,938), Fgfr4 (+34,204) | enhD (3X) |
| 14qB | Cts3 (−517,722), Zfp808 (−42,067) | enhD (7X) |
| 15qE1 | Dph3 (−32,316), Msmb (−24,101) | prom (2X), enhP (4X), enhD (7X) |
| 18qE4 | Ndufa6 (−10,978), Cyp2d22 (+14,991) | enhD (3X) |
| 19qA2 | Tmx3 (−672,104), Dok6 (−68,522) | prom (2X), enhP (3X), enhD (16X) |
| XqF3 | Gstp3 (−4429) | enhD (3X) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantziou, S.; Markopoulos, G.S. Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology. Biomedicines 2021, 9, 1742. https://doi.org/10.3390/biomedicines9111742
Mantziou S, Markopoulos GS. Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology. Biomedicines. 2021; 9(11):1742. https://doi.org/10.3390/biomedicines9111742
Chicago/Turabian StyleMantziou, Stefania, and Georgios S. Markopoulos. 2021. "Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology" Biomedicines 9, no. 11: 1742. https://doi.org/10.3390/biomedicines9111742
APA StyleMantziou, S., & Markopoulos, G. S. (2021). Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology. Biomedicines, 9(11), 1742. https://doi.org/10.3390/biomedicines9111742