
Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path


Abstract
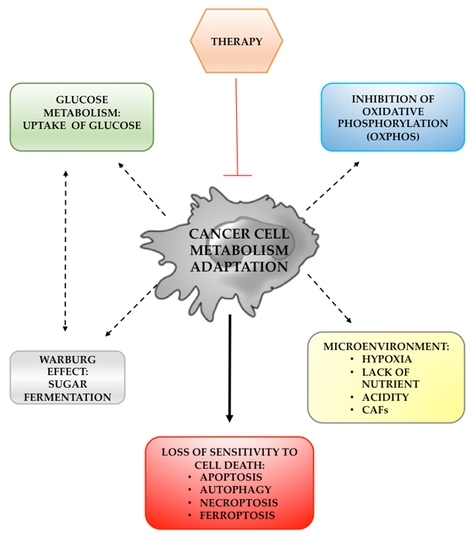
:
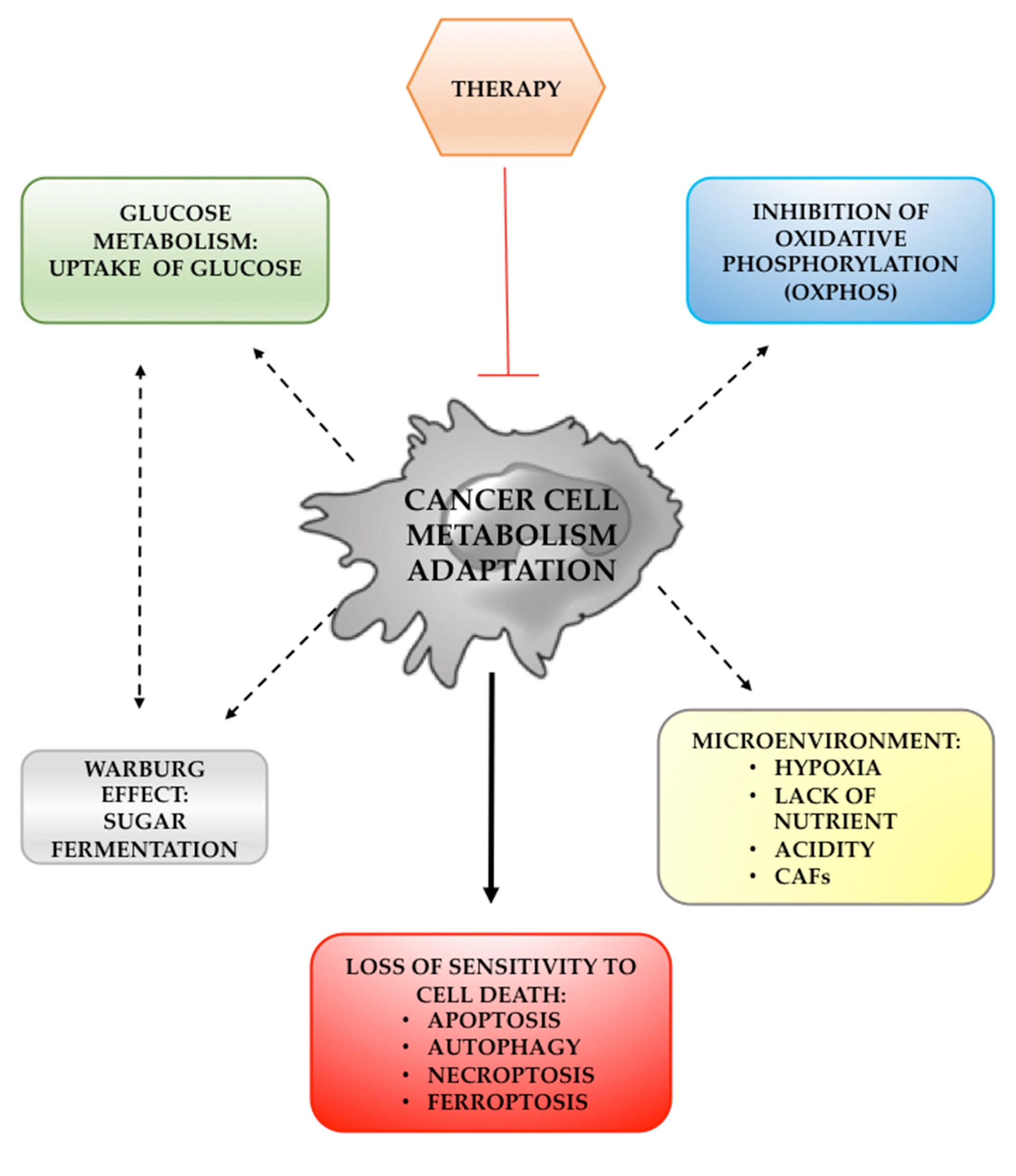{kind=link}
{kind=link}
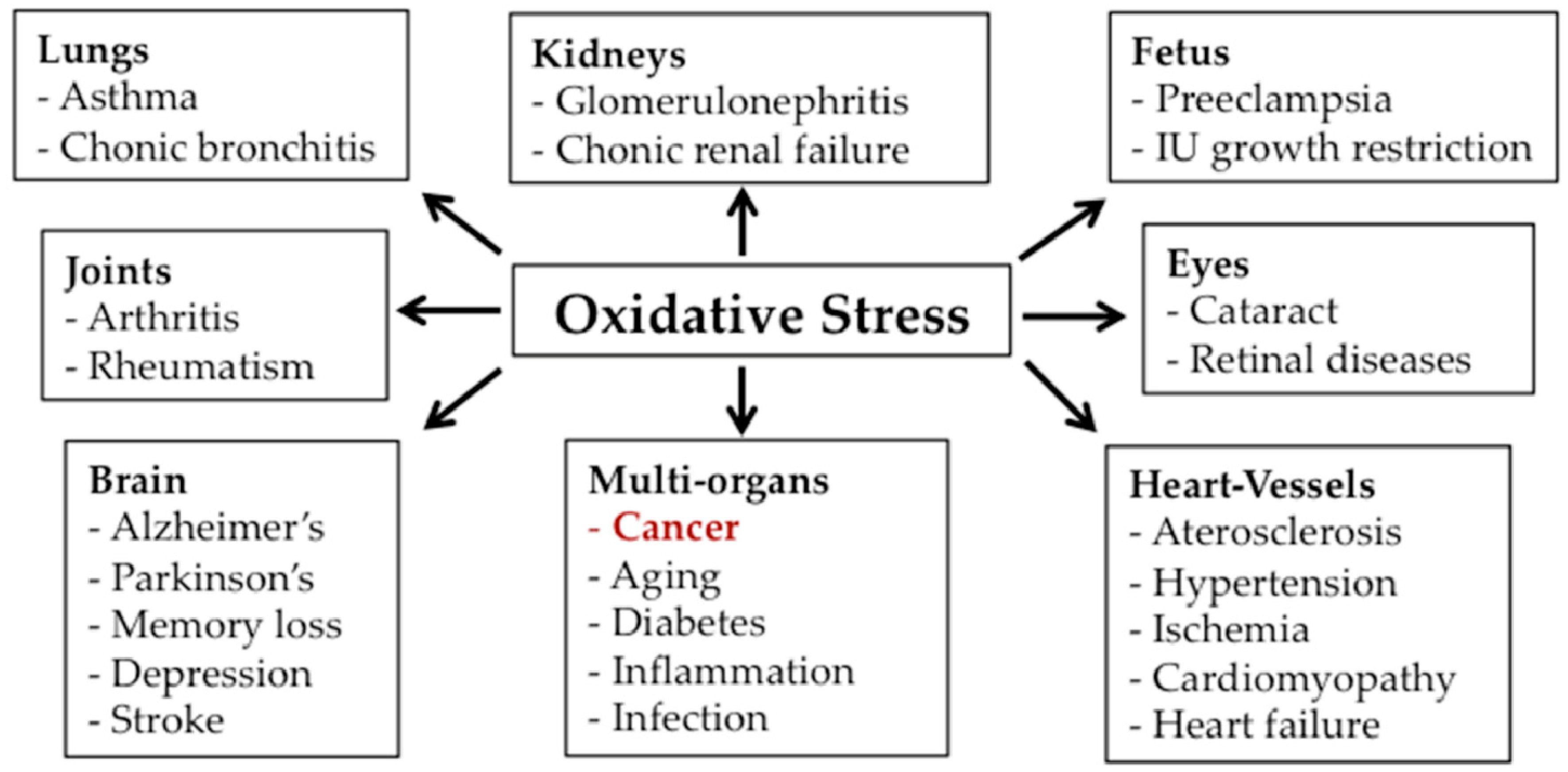{kind=link}
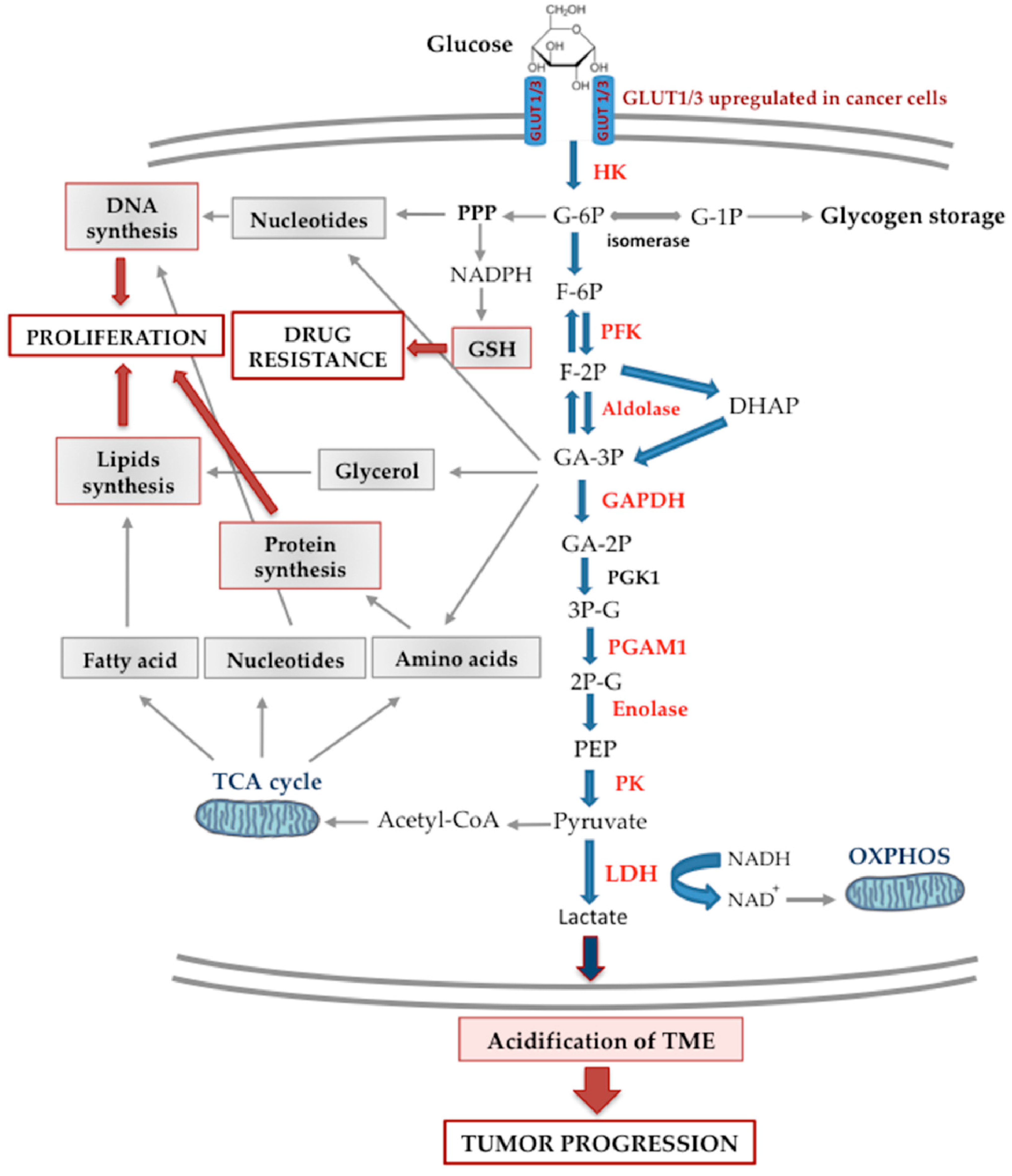{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mitochondria and Cancer Cell Metabolism
3. Glucose Metabolism Alteration in Cancer Cells
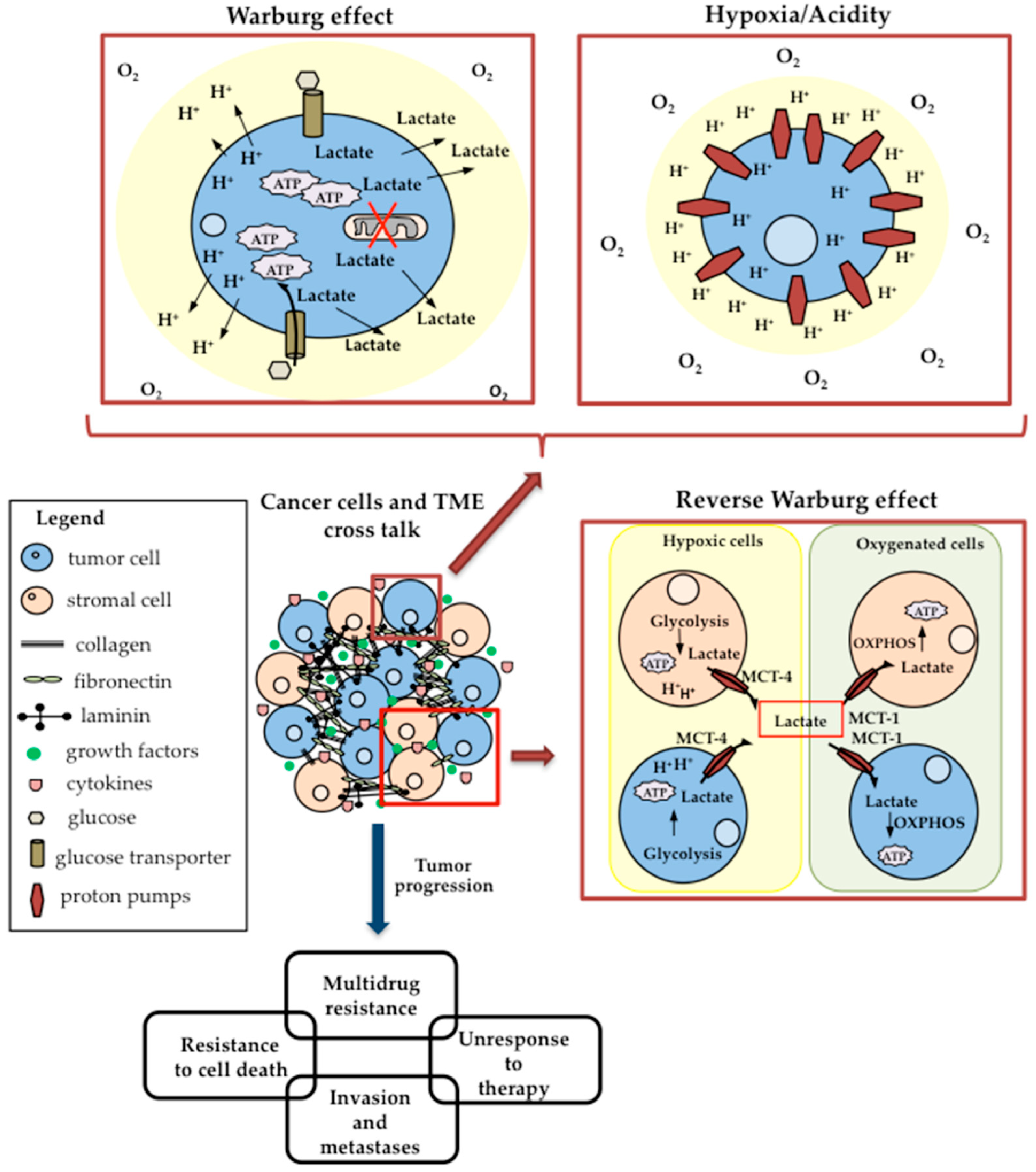
4. Warburg Effect and Glucose Metabolism Reprogramming
5. Tumor Microenvironment and Cancer Cell Metabolism
5.1. Hypoxia and Acidic pH of TME
5.2. Lactate Shuttle and the Reverse Warburg Effect
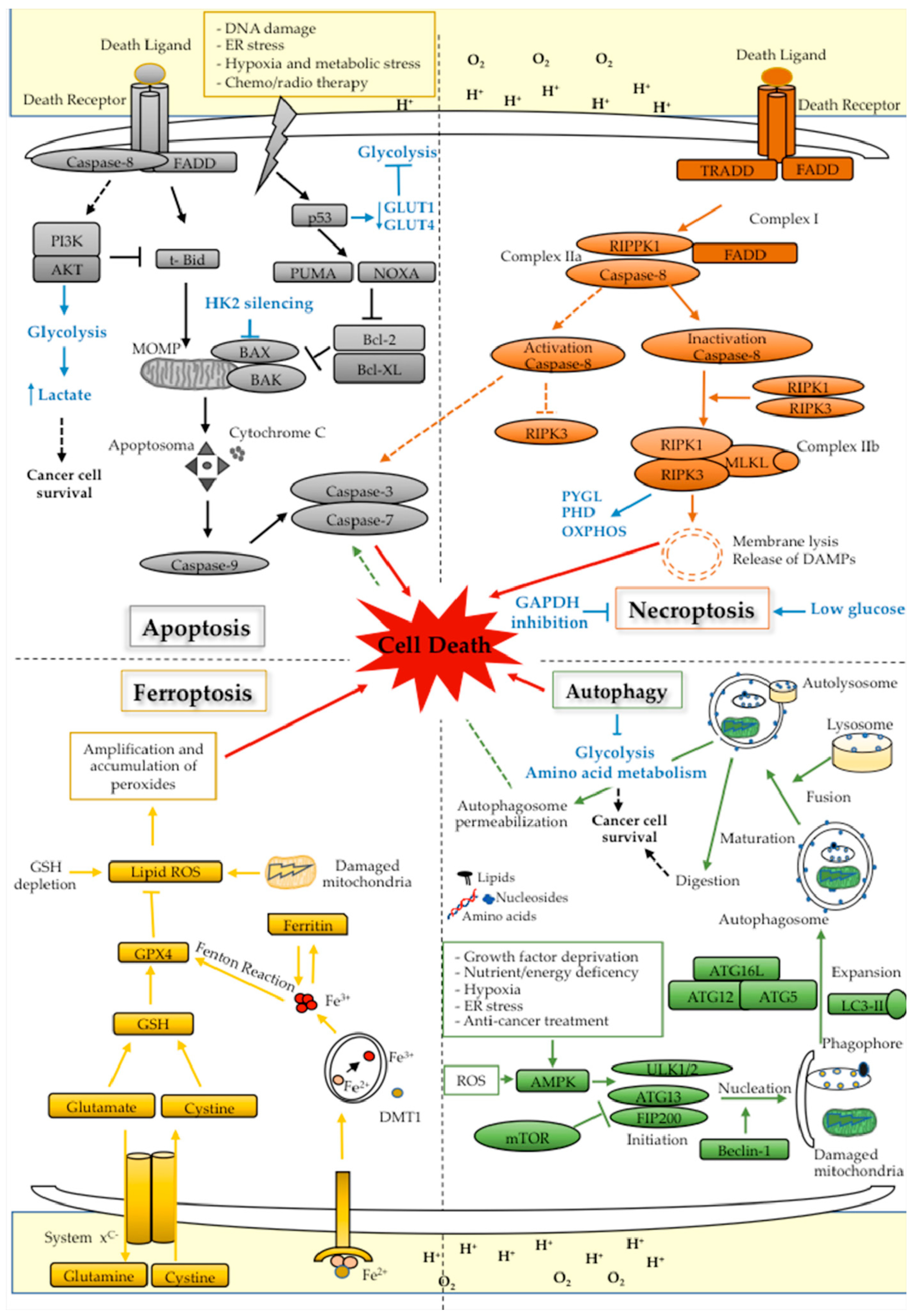
6. Cell Death and Metabolism in Cancer
6.1. Apoptosis
6.2. Autophagy
6.3. Necroptosis
6.4. Ferroptosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TCA | tricarboxylic acid |
| OXPHOS | oxidative phosphorylation |
| PPP | pentose phosphate pathway |
| ROS | reactive oxygen species |
| ATP | adenosine triphosphate |
| ADP | adenosine bisphosphate |
| ETC | electron transport chain |
| NADH | nicotinammina adenina dinucleotide |
| PGC1α | peroxisome proliferator activated receptor gamma coactivator-1α |
| HIFA | hypoxia inducible factor alpha |
| MnSOD | manganese-requiring mitochondrial enzyme |
| Trx | thioredoxin |
| TrxR | thioredoxin reductase |
| Prx3 | peroxidase 3 |
| NRF2 | nuclear factor erythroid- 2–related factor 2 |
| GSH | reduced glutathione |
| GSSG | oxydited glutathione |
| BPTES | bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-ylethyl sulfide |
| Bcl-2 | B-cell lymphoma 2 |
| 2DG | glycolysis inhibitor 2-Deoxy-D-glucose |
| GLUT | glucose transporters |
| G1P | glucose-1-phosphate |
| G6P | glucose-6-phosphate |
| HK2 | hexokinase 2 |
| PFK | phosphofructokinase |
| DHAP | dihydroxyacetone phosphate |
| GAPDH | glyceraldehyde-3-phosphate-dehydrogenase |
| PGAM1 | phosphoglycerate mutase 1 |
| PK | pyruvate kinase |
| LDH | lactate dehydrogenase |
| HIF-1α | hypoxia-inducible factor 1α |
| CoA | acetyl coenzyme A |
| TME | tumor microenvironment |
| CTCs | circulating tumor cells |
| cfDNA | cell-free DNA |
| HRE | hypoxia response element |
| MSR | magnetic resonance spectroscopy |
| V-ATPase | vacuolar H+-ATPase |
| NHE | Na+/H+ exchanger |
| MCTs | monocarboxylate transporters |
| CA-IX/XII | carbonic anhydrase IX/XII |
| NBCs | Na+/HCO3 co-transporters |
| PPIs | Proton Pumps Inhibitors |
| CAFs | cancer-associated fibroblasts |
| CAV1 | Caveolin-1 NF-kB |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| TIGAR | TP53 Induced Glycolysis and Apoptosis Regulator |
| TNF | tumor necrosis factor |
| MOMP | outer mitochondrial membrane |
| AKT | serine/threonine kinase |
| PUMA | p53 upregulated modulator of apoptosis |
| MCL-1 | Myeloid leukemia 1 |
| HCQ | hydroxychloroquine |
| Vps34 | class III phosphatylinositol 3-kinase |
| ULK1 | autophagy initiating kinases |
| TLRs | toll-like receptors |
| IFN | interferon |
| RIP | receptor-interacting protein |
| Nec-1 | Necrostatin-1 |
| TNFR1 | tumor necrosis factor receptor 1 |
| TRADD | TNFR-associated death domain |
| PYGL | glycogen phosphorylase |
| PDH | pyruvate dehydrogenase |
| PRRs | pattern recognition receptors |
| TCRs | T cell receptors |
| GPX4 | glutathione-dependent peroxidase 4 |
| DFO | chelator deferoxamine |
| NAC | the antioxidant N-acetyl-cysteine |
| PCL | Polygonatum cyrtonema lectins |
| TFR1 | iron specific receptor |
| Mfrn1 | mitoferrin 1 |
| Mfrn2 | mitoferrin 2 |
| VDACs | voltage-dependent anion channels |
| ER | endoplasmic reticulum |
| FADD | Fas Associated Via Death Domain |
| Bcl-XL | B-cell lymphoma-extra large |
| Bax | BCL2 Associated X |
| Bad | BCL2 Associated Agonist of Cell Death |
| tBid | truncated BH3 Interacting Domain Death Agonist |
| PI3K | Phosphoinositide 3-kinase |
| AKT | serine/threonine kinase |
| MLKL | Mixed lineage kinase domain-like protein |
| DAMPS | Damage-associated molecular patterns |
| AMPK | AMP-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| FLIP | Cellular FLICE-like inhibitory protein |
| LC3 | Microtubule-associated protein 1A/1B-light chain |
| DMT11 | Divalent metal transporter-1 |
References
- Vaghari-Tabari, M.; Ferns, G.A.; Qujeq, D.; Andevari, A.N.; Sabahi, Z.; Moein, S. Signaling, metabolism, and cancer: An important relationship for therapeutic intervention. J. Cell. Physiol. 2021, 236, 5512–5532. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Foo, B.J.; Eu, J.Q.; Hirpara, J.L.; Pervaiz, S. Interplay between Mitochondrial Metabolism and Cellular Redox State Dictates Cancer Cell Survival. Oxid. Med. Cell. Longev. 2021, 3, 1341604. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Zhang, F.; Li, D.; Li, C.; Li, H.; Jiang, Y.; Zhang, D. Overexpression of UQCRC2 is correlated with tumor progression and poor prognosis in colorectal cancer. Pathol. Res. Pract. 2018, 214, 1613–1620. [Google Scholar] [CrossRef]
- Gao, F.; Liu, Q.; Li, G.; Dong, F.; Qiu, M.; Lv, X.; Zhang, S.; Guo, Z. Identification of ubiquinol cytochrome c reductase hinge (UQCRH) as a potential diagnostic biomarker for lung adenocarcinoma. Open Biol. 2016, 6, 150256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Chen, X.; Li, X.; Luo, B. Inhibition of mitochondrial respiration overcomes hepatocellular carcinoma chemoresistance. Biochem. Biophys. Res. Commun. 2019, 508, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sànchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation- resistant pancreatic cancer cells depend on mithocondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellinga, T.T.; Borovski, T.; De Boer, V.C.J.; Fatrai, S.; Von Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.; Koester, J. SIRT1/PGC1α- Dependent Increase in Oxidative phosphorilation supports chemotherapy resistance of colon cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-fluorouacil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalcon, V.; Bindoli, A.; Rigobello, M.P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radic. Biol. Med. 2018, 127, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Thompson, M.A.; Tamayo, A.T.; Zuo, Z.; Lee, J.; Vega, F.; Ford, R.J.; Pham, L.V. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget 2012, 3, 314–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.Y.; Cantley, L.C.; DeNicola, G.M. NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response. In A Master Regulator of Oxidative Stress—The Transcription Factor Nrf2; IntechOpen: London, UK, 2016. [Google Scholar]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic targeting synergizes with MAPK inhibition and delays drug resistance in melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palchaudhuri, R.; Lambrecht, M.J.; Botham, R.C.; Partlow, K.C.; van Ham, T.J.; Putt, K.S.; Nguyen, L.T.; Kim, S.H.; Peterson, R.T.; Fan, T.M.; et al. A Small Molecule that Induces Intrinsic Pathway Apoptosis with Unparalleled Speed. Cell Rep. 2015, 13, 2027–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffez, H.; Taha, H.; Farrag, N.S.; Amin, A.M.; Hassan, Z.A. Biological Screening and Radiolabeling of Raptinal as a Potential Anticancer Novel Drug in Hepatocellular Carcinoma Model. Eur. J. Pharm. Sci. 2021, 158, 105653. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Vona, R.; Mileo, A.M.; Matarrese, P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers 2021, 13, 5812. [Google Scholar] [CrossRef]
- Caino, M.C.; Altieri, D.C. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, D.C. Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 2017, 117, 301–305. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, A.; Molt, M.; Uribe, E.; Salas, M. Glut 1 in Cancer Cells and the Inhibitory Action of Resveratrol as A Potential Therapeutic Strategy. Int. J. Mol. Sci. 2019, 20, 3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TeSlaa, T.; Bartman, C.R.; Jankowski, C.S.R.; Zhang, Z.; Xu, X.; Xing, X.; Wang, L.; Lu, W.; Hui, S.; Rabinowitz, J.D. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab. 2021, 33, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen Metabolism in Cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.E.; Mayfield, A.C.; Haas, J.A. Heart muscle hexokinase: Subcellular distribution and inhibition by glucose 6-phosphate. Mol. Pharmacol. 1966, 2, 393–405. [Google Scholar] [PubMed]
- Garcia, N.S.; Guedes, C.R.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Cur. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Mehta, R.; Sonavane, M.; Migaud, M.E.; Gassman, N.R. Exogenous exposure to dihydroxyacetone mimics high fructose induced oxidative stress and mitochondrial dysfunction. Environ. Mol. Mutagen 2021, 62, 185–202. [Google Scholar] [CrossRef]
- Gupta, V.; Bamezai, R.N.K. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP Proteins Controlled by C-Myc Deregulate Pyruvate Kinase mRNA Splicing in Cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Le, H. Dual roles of PKM2 in cancer metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, X. Phosphoglycerate Mutase 1: Its Glycolytic and Non-Glycolytic Roles in Tumor Malignant Behaviors and Potential Therapeutic Significance. OncoTargets Ther. 2020, 13, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1 through NF-kB activation. Mol. Cancer 2016, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget 2017, 9, 4020–4033. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, K.; Nakamura, Y.; Sakata, K.; Fujimori, K.; Ohkubo, M.; Sawada, K.; Sakiyama, S. Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res. 1987, 47, 5616–5619. [Google Scholar] [PubMed]
- Zancan, P.; Sola-Penna, M.; Furtado, C.M.; da Silva, D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Mol. Genet. Metab. 2010, 100, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xiao, Z.; Chen, Z.; Zhang, X.; Li, J.; Wu, X.; Li, X.; Yi, H.; Li, M.; Zhu, G.; et al. Proteome analysis of human lung squamous carcinoma. Proteomics 2006, 6, 547–558. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ hell. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Ca, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From Warburg effect to Reverse Warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49, 24S–42S. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Mao, X.; Mehta, M.; Cui, J.; Zhang, C.; Mao, F.; Xu, Y. Elucidation of how cancer cells avoid acidosis through comparative transcriptomic data analysis. PLoS ONE 2013, 8, e71177. [Google Scholar] [CrossRef] [Green Version]
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.; Carcel-Trullols, J.; Xie, C.; Evans, T.T.; Mizumachi, T.; Higuchi, M. Induction of acquired resistance to antiestrogen by reversible mitochondrial DNA depletion in breast cancer cell line. Int. J. Cancer 2008, 122, 1506–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraresi, R.; Troiano, L.; Pinti, M.; Roat, E.; Lugli, E.; Quaglino, D.; Taverna, D.; Bellizzi, D.; Passarino, G.; Cossarizza, A. Resistance of mtDNA-depleted cells to apoptosis. Cytom. A 2008, 73, 528–537. [Google Scholar] [CrossRef]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; Seidi, K.; Banimohamad-Shotorbani, B.; Jahanban-Esfahlan, A.; Yousefi, B. Combination of nanotechnology with vascular targeting agents for effective cancer therapy. J. Cell Physiol. 2017, 233, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; Seidi, K.; Zarghami, N. Tumor vascular infarction: Prospects and challenges. Int. J. Hematol. 2017, 105, 244–256. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, K.T.; Alazzech, A.; Werb, Z.; Weaver, V.M. Leveraging microenvironmental synthetic lethalities to treat cancer. J. Clin. Investig. 2021, 131, e143765. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Mattia, G.; Pagano, M.T.; Pontecorvi, G.; Ortona, E.; Malorni, W.; Carè, A. The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy. Cancers 2021, 13, 3287. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef] [Green Version]
- Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Supuran, C.T.; Fais, S. Rethinking the Combination of Proton Exchanger Inhibitors in Cancer Therapy. Metabolites 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.C.; Rouhi, P.; Zois, C.E.; Haider, S.; Li, J.L.; Kessler, B.M.; Cao, Y.; Harris, A.L. Hypoxic regulation of RIOK3 is a major mechanism for cancer cell invasion and metastasis. Oncogene 2015, 34, 4713–4722. [Google Scholar]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia Inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covello, K.L.; Simon, M.C. HIFs, hypoxia, and vascular development. Curr. Top. Dev. Biol. 2004, 62, 37–54. [Google Scholar] [PubMed]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [Green Version]
- Beasley, N.J.; Leek, R.; Alam, M.; Turley, H.; Cox, G.J.; Gatter, K.; Millard, P.; Fuggle, S.; Harris, A.L. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: Relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res. 2002, 62, 2493–2497. [Google Scholar] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.C.; Harris, A.L. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volm, M.; Koomagi, R. Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res. 2000, 20, 1527–1533. [Google Scholar]
- Huang, M.; Yang, L.; Peng, X.; Wei, S.; Fan, Q.; Yang, S.; Li, X.; Li, B.; Jin, H.; Wu, B.; et al. Autonomous glucose metabolic reprogramming of tumour cells under hypoxia: Opportunities for targeted therapy. J. Exp. Clin. Cancer Res. 2020, 39, 185. [Google Scholar] [CrossRef]
- Shen, G.M.; Zhang, F.L.; Liu, X.L.; Zhang, J.W. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett. 2010, 584, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akanji, M.A.; Rotimi, D.; Adeyemi, O.S. Hypoxia-inducible factors as an alternative source of treatment strategy for Cancer. Oxid. Med. Cell. Longev. 2019, 2019, 8547846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.; Zhao, X.; Wang, X.; Zhao, Y.; Li, Y.; Zhao, R.; Cheng, K.; Li, Y.; Han, X.; Zheng, X.; et al. Targeted co-delivery of the Iron Chelator Deferoxamine and a HIF1alpha inhibitor impairs pancreatic tumor growth. ACS Nano 2019, 13, 2176–2189. [Google Scholar]
- Liu, Y.X.; Feng, J.Y.; Sun, M.M.; Liu, B.W.; Yang, G.; Bu, Y.N.; Zhao, M.; Wang, T.J.; Zhang, W.Y.; Yuan, H.F.; et al. Aspirin inhibits the proliferation of hepatoma cells through controlling GLUT1-mediated glucose metabolism. Acta Pharmacol. Sin. 2019, 40, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumarola, C.; Cretella, D.; La Monica, S.; Bonelli, M.A.; Alfieri, R.; Caffarra, C.; Quaini, F.; Madeddu, D.; Falco, A.; Cavazzoni, A.; et al. Enhancement of the antitumor activity of FGFR1 inhibition in squamous cell lung cancer by targeting downstream signaling involved in glucose metabolism. Oncotarget 2017, 8, 91841–91859. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, T.A.; Farias, L.C.; Santos, E.S.; de Carvalho Fraga, C.A.; Orsini, L.A.; de Freitas Teles, L.; Feltenberger, J.D.; de Jesus, S.F.; de Souza, M.G.; Santos, S.H.; et al. Metformin increases PDH and suppresses HIF-1alpha under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget 2016, 7, 55057–55068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Li, X.; Luo, Z.; Liu, J.; Fan, Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol. Cancer Ther. 2013, 12, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.D.; Tyler, T.J.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019, 20, e46557. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of a tumor chemoresistance: Proton Pump Inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updat. 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Canitano, A.; Iessi, E.; Spugnini, E.P.; Federici, C.; Fais, S. Proton Pump Inhibitors induce a caspase-independent antitumor effect against human multiple myeloma. Cancer Lett. 2016, 376, 278–283. [Google Scholar] [CrossRef]
- Anemone, A.; Consolino, L.; Conti, L.; Irrera, P.; Hsu, M.Y.; Villano, D.; Dastrù, W.; Porporato, P.E.; Cavallo, F.; Longo, D.L. Tumor acidosis evaluated in vivo by MRI-CEST pH imaging reveals breast cancer metastatic potential. Br. J. Cancer 2021, 124, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Aunet, S.; Di Pompo, G.; Lemma, S.; Baldini, N. Cause and effect of microenvironmental acidosis on bone metastatic. Cancer Metastasis Rev. 2019, 38, 133–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wike-Hooney, J.L.; Haven, J.; Reinhold, H.S. The relevance of tumor pH to the treatment of malignant disease. Radiother. Oncol. 1984, 2, 343–366. [Google Scholar] [CrossRef]
- Gallaghe, F.A.; Kettunen, M.I.; Day, S.E.; Hu, D.E.; Ardenkjaer-Larsen, J.M.; Zandt, R.; Jensen, P.R.; Karlsson, M.; Golman, K.; Lerche, M.M.; et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature 2008, 453, 940–943. [Google Scholar] [CrossRef]
- Fais, S.; Venturi, G.; Gatenby, B. Microenvironmental acidosis in carcinogenesis and metastases: New strategies in prevention and therapy. Cancer Met. Rev. 2014, 33, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lu, W.; Garcia-Prieto, C.; Huang, P. The Warburg effect and its cancer therapeutic implications. J. Bioenerg. Biomembr. 2007, 39, 267–274. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, A.G.; Garcìa, S.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [Green Version]
- Fais, S.; De Milito, A.; You, H.; Qin, W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007, 67, 10627–10630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H + ATPase (V-ATPase): From proton pump to signaling complex in health and disease. Am J. Physiol. Cell Physiol. 2020, 320, C392–C414. [Google Scholar] [CrossRef] [PubMed]
- Svastová, E.; Hulíková, A.; Rafajová, M.; Zat’ovicová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatenko, A.; Olesen, C.W.; Boedtkjer, E.; Pedersen, S.F. Regulation and roles of bicarbonate transporters in cancer. Front. Physiol. 2014, 5, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sennoune, S.R.; Martinez-Zaguilan, R. Plasmalemmal vacuolar H+-ATPases in angiogenesis, diabetes and cancer. J. Bioenerg. Biomembr. 2007, 39, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galenkamp, K.M.O.; Sosicka, P.; Jung, M.; Recouvreux, M.V.; Zhang, Y.; Moldenhauer, M.R.; Brandi, G.; Freeze, H.H.; Commisso, C. Golgi acidification by NHE7 regulates cytosolic pH homeostasis in pancreatic cancer cells. Cancer Discov. 2020, 10, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Luciani, F.; Spada, M.; De Milito, A.; Molinari, A.; Rivoltini, L.; Montinaro, A.; Marra, M.; Lugini, L.; Logozzi, M.; Lozupone, F.; et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Baldi, A.; Buglioni, S.; Carocci, F.; de Bazzichini, G.M.; Betti, G.; Pantaleo, I.; Menicagli, F.; Citro, G.; Fais, S. Lansoprazole as a rescue agent in chemoresistant tumors: A phase I/II study in companion animals with spontaneously occurring tumors. J. Transl. Med. 2011, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.; Perut, F.; Fagioli, F.; Brach Del Prever, A.; Meazza, C.; Parafioriti, A.; Picci, P.; Gambarotti, M.; Avnet, S.; Baldini, N.; et al. Proton pump inhibitor chemosensitization in human osteosarcoma: From the bench to the patients’ bed. J. Transl. Med. 2013, 11, 268. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.Y.; Zhang, J.; Wang, J.L.; Sun, S.; Wang, Z.H.; Wang, L.P.; Zhang, Q.L.; Lv, F.F.; Cao, E.Y.; Shao, Z.M.; et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 85. [Google Scholar] [CrossRef] [Green Version]
- Falcone, R.; Roberto, M.; D’Antonio, C.; Romiti, A.; Milano, A.; Onesti, C.E.; Marchetti, P.; Fais, S. High-doses of proton pumps inhibitors in refractory gastro-intestinal cancer: A case series and the state of art. Dig. Liver Dis. 2016, 48, 1503–1505. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Milano, A.; D’Antonio, C.; Romiti, A.; Falcone, R.; Roberto, M.; Fais, S. Association between proton pump inhibitors and metronomic capecitabine as salvage treatment for patients with advanced gastrointestinal tumors: A randomized phase II trial. Clin. Colorectal Cancer 2016, 15, 377–380. [Google Scholar] [CrossRef]
- De Milito, A.; Iessi, E.; Logozzi, M.; Lozupone, F.; Spada, M.; Marino, M.L.; Federici, C.; Perdicchio, M.; Matarrese, P.; Lugini, L.; et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007, 67, 5408–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef]
- Feng, S.; Zheng, Z.; Feng, L.; Yang, L.; Chen, Z.; Lin, Y.; Gao, Y.; Chen, Y. Proton pump inhibitor pantoprazole inhibits the proliferation, self-renewal and chemoresistance of gastric cancer stem cells via the EMT/-catenin pathways. Oncol. Rep. 2016, 36, 3207–3214. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Jeon, H.K.; Hong, J.E.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Lee, S.H.; Yoon, G.; Kim, W.Y.; Do, I.G. Proton pump inhibitors enhance the effects of cytotoxic agents in chemoresistant epithelial ovarian carcinoma. Oncotarget 2015, 6, 35040–35050. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Ge, Z.; Yang, X.; Luo, Q.; Wang, C.; You, H.; Ge, T.; Deng, Y.; Lin, H.; Cui, Y.; et al. Hepatic stellate cells activated by acidic tumor microenvironment promote the metastasis of hepatocellular carcinoma via osteopontin. Cancer Lett. 2015, 356, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Arranz, J.L.; Polo Orozco, J.D.; Rauch, C.; Fais, S.; Cardone, R.A.; Reshkin, S.J. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—An integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J. Transl. Med. 2013, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, C.; Schwab, A.; et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshkin, S.J.; Bellizzi, A.; Cardone, R.A.; Tommasino, M.; Casavola, V.; Paradiso, A. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin. Cancer Res. 2003, 9, 2366–2373. [Google Scholar] [PubMed]
- Counillon, L.; Bouret, Y.; Marchiq, I.; Pouyssegur, J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 2016, 1863, 2465–2480. [Google Scholar] [CrossRef]
- Aredia, F.; Scovassi, A.I. Multiple effects of intracellular pH modulation in cancer cells. Cancer Cell. Microenv. 2014, 1, 72–79. [Google Scholar]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell. Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, V.; Santamaria, G.; Torriglia, A.; Aredia, F.; Scovassi, A.I.; Bottiroli, G.; Croce, A.C. Fluorescence properties of the Na+/H+ exchanger inhibitor HMA (5-(N, Nhexamethylene) amiloride) are modulated by intracellular pH. Eur. J. Histochem. 2012, 56, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aredia, F.; Giansanti, V.; Mazzini, G.; Savio, M.; Ortiz, L.M.G.; Jaadane, I.; Zaffaroni, N.; Forlino, A.; Torriglia, A.; Ivana Scovassi, A. Multiple effects of the Na(+)/H (+) antiporter inhibitor HMA on cancer cells. Apoptosis 2013, 18, 1586–1598. [Google Scholar] [CrossRef]
- Aredia, F.; Czaplinski, S.; Fulda, S.; Scovassi, A.I. Molecular features of the cytotoxicity of an NHE inhibitor: Evidence of mitochondrial alterations, ROS overproduction and DNA damage. BMC Cancer 2016, 16, 851. [Google Scholar] [CrossRef] [Green Version]
- Rowson-Hodel, A.R.; Berg, A.L.; Wald, J.H.; Hatakeyama, J.; VanderVorst, K.; Curiel, D.A.; Leon, L.J.; Sweeney, C.; Carraway, K.L. Hexamethylene amiloride engages a novel reactive oxygen pecies- and lysosome-dependent programmed necrotic mechanism to selectively target breast cancer cells. Cancer Lett. 2016, 375, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonveaux, P.; Vègran, F.; Schroeder, T.; Wrgin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todenhöfer, T.; Seiler, R.; Stewart, C.; Moskalev, I.; Gao, J.; Ladhar, S.; Kamjabi, A.; Al Nakouzi, N.; Hayashi, T.; Choi, S.; et al. Selective Inhibition of the Lactate Transporter MCT4 Reduces Growth of Invasive Bladder Cancer. Mol. Cancer Ther. 2018, 17, 2746–2755. [Google Scholar] [CrossRef] [Green Version]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzene sulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, P.C.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J.; et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2, e92928. [Google Scholar] [CrossRef] [PubMed]
- Wartenber, M.; Ling, F.C.; Muschen, M.; Klein, F.; Acker, H.; Gassmann, M.; Petrat, K.; Pütz, V.; Hescheler, J.; Sauer, H. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. FASEB J. 2003, 17, 503–505. [Google Scholar]
- Welch, H.G. The heterogeneity of cancer. Breast Cancer Res. Treat. 2018, 169, 207–208. [Google Scholar] [CrossRef] [Green Version]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Hardee, M.E.; Dewhirst, M.W.; Agarwal, N.; Sorg, B.S. Novel imaging provides new insights into mechanisms of oxygen transport in tumors. Curr. Mol. Med. 2009, 9, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Yoon, J.-H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumours. Cell Cycle 2011, 20, 10. [Google Scholar]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumour stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Quanz, M.; Bender, E.; Kopitz, C.; Grünewald, S.; Schlicker, A.; Schwede, W.; Eheim, A.; Toschi, L.; Neuhaus, R.; Richter, C.; et al. Preclinical efficacy of the novel monocarboxylate transporter 1 inhibitor BAY-8002 and associated markers of resistance. Mol. Cancer Ther. 2018, 17, 2285–2296. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.B.; Solass, W.; Archid, R.; Weinreich, F.J.; Königsrainer, A.; Reymond, M.A. Resistance to anoikis in transcoelomic shedding: The role of glycolytic enzymes. Pleura Peritoneum 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Harris, A.L. Pyruvate Dehydrogenase and Pyruvate Dehydrogenase Kinase Expression in Non Small Cell Lung Cancer and Tumor-Associated Stroma. Neoplasia 2005, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-B/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, C.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Tanowitz, H.B.; Pestell, R.G.; Sotgia, G.; Lisanti, M.P. Metabolic reprogramming and two-compartment tumor metabolism: Opposing role(s) of HIF1 alpha and HIF2alpha in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle 2012, 11, 3280–3289. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.H.; Domingo-Vidal, M.; Roche, M.; Bartrons, R.; Caro, J.; Martinez-Outschoom, U. TP53- inducible Glycolysis and Apoptosis Regular (TIGAR) metabolically reprograms carcinoma and stromal cells in breast cancer. J. Biol. Chem. 2016, 291, 26291–26303. [Google Scholar] [CrossRef] [Green Version]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 2012, 87, 33436–33446. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xie, W.; Li, Q.; Zhang, Y.; Zhang, J.; Zhao, X.; Huang, G. TIGAR is correlated with maximal standardized uptake value on FDG-PET and survival in non-small cell lung cancer. PLoS ONE 2013, 8, e80576. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Li, J.; Hong, M.; Zhu, Y.; Zhao, H.; Xie, Y.; Huang, J.; Lian, Y.; Li, Y.; Wang, S.; et al. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J. Hematol. Oncol. 2016, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Attucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendation of the Nomenclature Committee 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Wolpaw, A.; Dang, C.V. Exploting metabolic Vulnerabilities of Cancer with Precision and Accuracy. Trends Cell Biol. 2018, 28, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic regulation of Apoptosis in Cancer. Int. Rev. Cell Mol. Bio. 2016, 327, 43–87. [Google Scholar]
- Thornberry, N.A.; Lazbenik, Y. Caspasaes: Enemies within. Science 2018, 281, 1312–1316. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Cawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosi sas anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Wang, J. Hypoxia selection of death-resistant cells. A role for Bcl-X(L). J. Biol. Chem. 2004, 279, 9215–9221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism: A key to apoptosis resistance? Cell Cycle 2007, 6, 790–792. [Google Scholar] [CrossRef] [Green Version]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncongene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Brosius, F.C. Glucose uptake and glycolysis reduce hypoxia-induced apoptosis in cultured neonatal rat cardiac myocytes. J. Biol. Chem. 1999, 274, 12567–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Patra, K.C.; Hay, N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. eLife 2018, 7, e32213. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of henokinase II inihibts Bax-induced cytrochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.N.; Yu, B.B.; Li, J.L.; Guo, R.; Zhang, L.C.; Sun, L.K.; Liu, Y.N.; Li, Y. Zinc and p53 disrupt mitochondrial binding of HK2 by phosphorylating VDAC1. Exp. Cell Res. 2019, 374, 249–258. [Google Scholar] [CrossRef]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis-the p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT kinase in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [PubMed]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell. 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coloff, J.L.; Macintyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Altman, B.; Coloff, J.L.; Herman, C.E.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Dimascio, L.N.; Ilkayeva, O.; Kelekar, A.; et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol. Cell Biol. 2007, 27, 4328–4339. [Google Scholar] [CrossRef] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Majewski, N.; Nogueira, V.; Robey, R.B.; Hay, N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell Biol. 2004, 24, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.-H.; Lee, J.H.; Park, J.W.; Quach, C.H.T.; Moon, S.H.; Cho, Y.-S.; Lee, K.-H. Resveratrol-loaded polymeric nanoparticles suppress glucose metabolism and tumor growth in vitro and in vivo. Int. J. Pharm. 2015, 478, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Fouad, M.A.; Agha, A.M.; Al Merzabani, M.M.; Shouman, S.A. Resveratrol inhibits proliferation, angiogenesis and induces apoptosis in colon cancer cells. Hum. Exp. Toxicol. 2013, 32, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, X.; Li, N.; Liu, H.; Dong, Q.; Zhang, J.; Yang, C.; Liu, Y.; Liang, Q.; Zhang, S.; et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp. Cell Res. 2016, 349, 320–327. [Google Scholar] [CrossRef]
- Kueck, A.; Opipari, A.W.; Griffith, K.A.; Tan, L.; Choi, M.; Huang, J.; Wahl, H.; Liu, J.R. Resveratrol inhibits glucose metabolism in human ovarian cancer cells. Gynecol. Oncol. 2007, 107, 450–457. [Google Scholar] [CrossRef]
- Nakashima, S.; Hiraku, Y.; Tada-Oikawa, S.; Hishita, T.; Gabazza, E.C.; Tamaki, S.; Imoto, I.; Adachi, Y.; Kawanishi, S. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J. Biochem. 2003, 134, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Morimura, T.; Fujita, K.; Akita, M.; Nagashima, M.; Satomi, A. The proton pump inhibitor inhibits cell growth and induces apoptosis in human hepatoblastoma. Pediatr. Surg. Int. 2008, 24, 1087–1094. [Google Scholar] [CrossRef]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, J.M.; Gabello, M.; Murray, L.J.; Farrell, C.P.; Bellows, J.; Wolov, K.R.; Kearney, K.R.; Rudolph, D.; Thornton, J.J. Proton pump inhibitors: Actions and reactions. Drug Discov. Today 2009, 14, 647–660. [Google Scholar] [CrossRef]
- Shankar Babu, M.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar] [CrossRef] [Green Version]
- De Grandis, R.A.; Oliveira, K.M.; Guedes, A.P.M.; Dos Santos, P.W.S.; Aissa, A.F.; Batista, A.A.; Pavan, F.R. A Novel Ruthenium(II) Complex With Lapachol Induces G2/M Phase Arrest Through Aurora-B Kinase Down-Regulation and ROS-Mediated Apoptosis in Human Prostate Adenocarcinoma. Cells Front. Oncol. 2021, 11, 682968. [Google Scholar] [CrossRef]
- Zu, X.; Xie, X.; Zhang, Y.; Liu, K.; Bode, A.M.; Dong, Z.; Kim, D.J. Lapachol is a novel ribosomal protein S6 kinase 2 inhibitor that suppresses growth and induces intrinsic apoptosis in esophageal squamous cell carcinoma cells. Phytother. Res. 2019, 33, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Marques, L.B.; Ottoni, F.M.; Pinto, M.C.X.; Ribeiro, J.M.; de Sousa, F.S.; Weinlich, R.; de Victo, N.C.; Kisitu, J.; Holzer, A.K.; Leist, M.; et al. Lapachol acetylglycosylation enhances its cytotoxic and pro-apoptotic activities in HL60 cells. Toxicol. In Vitro 2020, 65, 104772. [Google Scholar] [CrossRef]
- Zhang, H.; Du, X.; Sun, T.T.; Wang, C.L.; Li, Y.; Wu, S.Z. Lectin PCL inhibits the Warburg effect of PC3 cells by combining with EGFR and inhibiting HK2. Oncol. Rep. 2017, 37, 1765–1771. [Google Scholar] [CrossRef]
- Shin, N.; Lee, H.J.; Sim, D.Y.; Im, E.; Park, J.E.; Park, W.Y.; Cho, A.R.; Shim, B.S.; Kim, S.H. Apoptotic effect of compound K in hepatocellular carcinoma cells via inhibition of glycolysis and Akt/mTOR/c-Myc signaling. Phytothe. Res. 2021, 35, 3812–3820. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Ying, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpandel, V.; Seling, M.K.; Ferrone, C.R.; et al. Transcriptional control of the autophagylysosome system in pancreatic cancer. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.Y. Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 2016, 3, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Mosquera, L.; Diogo, C.V.; Yambire, K.F.; Santos, G.L.; Luna Sanchez, M.; Benit, P.; Rustin, P.; Lopez, L.C.; Milosevic, I.; Raimundo, N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep. 2017, 7, 45076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [Green Version]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic resposne. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Mitter, S.K.; Qi, X.; Beli, E.; Rao, H.V.; Ding, J.; Ip, C.S.; Gu, H.; Akin, D.; Dunn, W.A., Jr.; et al. Oxidative stress-mediated NFkB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS ONE 2017, 12, e0171940. [Google Scholar]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase i trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, B.; El-Ahmad, Y.; Filoche-Romme, B.; Dureuil, C.; Fassy, F.; Abecassis, P.Y.; Mathieu, M.; Bertrand, T.; Benard, T.; Barrière, C.; et al. Discovery of (2s)-8-[(3r)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)- 3,4-dihydro-2h-pyrimido[1,2-a]pyrimidin-6-one: A novel potent and selective inhibitor of vps34 for the treatment of solid tumors. J. Med. Chem. 2015, 58, 376–400. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ulk1 and identification of ulk1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, S.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Veken, P.V.D.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Zahedi, S.F.; Fitzwalter, B.E.; Morin, A.; Grob, S.; Desmarais, M.; Nellan, A.; Green, A.L.; Vibhakar, R.; Hankinson, T.C.; Foreman, N.K.; et al. Effect of early stage autophagy inhibition in BRAFV600E autophagy dependent brain tumor cells. Cell Death Dis. 2019, 10, 679. [Google Scholar] [CrossRef] [Green Version]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, 215152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xipell, E.; Gonzalez-Huarriz, M.; de Irujo, J.J.M.; García-Garzón, A.; Lang, F.F.; Jiang, H.; Fueyo, J.; Gomez-Manzano, C.; Alonso, M.M.; Alonso, M.M. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget 2016, 7, 30626–30641. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, S.; Cheng, X.; Wu, J.; Wu, L.; Wang, Y.; Wang, X.; Bao, J.; Yu, H. Curcumin induces autophagic cell death in human thyroid cancer cells. Toxicol. In Vitro 2021, 78, 105254. [Google Scholar] [CrossRef]
- Shao, C.; Wu, J.; Han, S.; Liu, Y.; Su, Z.; Zhu, H.L.; Liu, H.K.; Qian, Y. Biotinylated curcumin as a novel chemosensitizer enhances naphthalimide-induced autophagic cell death in breast cancer cells. Eur. J. Med. Chem. 2021, 228, 114029. [Google Scholar] [CrossRef]
- Hoshiko, T.; Kubota, Y.; Onodera, R.; Higashi, T.; Yokoo, M.; Motoyama, K.; Kimura, S. Folic Acid-Appended Hydroxypropyl-β-Cyclodextrin Exhibits Potent Antitumor Activity in Chronic Myeloid Leukemia Cells via Autophagic Cell Death. Cancers 2021, 13, 5413. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, K.; Haitao, S.; Yao, X.; Sun, Q.; Chen, G. Necroptosis: A novel manner of cell death, associated with stroke (Review). Int. J. Mol. Med. 2017, 41, 624–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikseresht, S.; Khodagholi, F.; Ahmadiani, A. Protective effects of ex-527 on cerebral ischemia-reperfusion injury through necroptosis signaling pathway attenuation. J. Cell Physiol. 2019, 234, 1816–1826. [Google Scholar] [CrossRef]
- Duriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Zhang, W.; Yang, T.; He, S.D. Complex roles of necroptosis in cancer. J. Zhejiang Univ. Sci. B 2019, 20, 399–413. [Google Scholar] [CrossRef]
- Qui, X.; Ma, D.; Tan, Y.X.; Wang, H.Y.; Cai, Z. The role of necroptosis in cancer: A double-edged sword? Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 259–266. [Google Scholar]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF- induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kuo, W.T.; Huang, Y.C.; Lee, T.C.; Yu, L.C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.R.; Xiang, S.; Song, Z.; Wu, M. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J. 2017, 36, 3483–3500. [Google Scholar] [CrossRef]
- McCaig, W.D.; Patel, P.S.; Sosunov, S.A.; Shakerley, N.L.; Smiraglia, T.A.; Craft, M.M.; Walker, K.M.; Deragon, M.A.; Ten, V.S.; LaRocca, T.J. Hyperglycemia potentiates a shift from apoptosis to RIP1-dependent necroptosis. Cell Death Discov. 2018, 4, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.V.; Chen, H.C.; Walsh, C.M. Necroptotic signaling in adaptive and innate immunity. Semin. Cell Dev. Biol. 2014, 35, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Mohammadinejad, R.; Tavakol, S.; Ahmadi, Z.; Roomiani, S.; Katebi, M. Autophagy, anoikis, ferroptosis, necroptosis, and endoplasmatic reticulum stress: Potential applications in melanoma therapy. J. Cell Physiol. 2019, 234, 19471–19479. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Jiang, X. To eat or not to eat—The metabolic flavor of ferroptosis. Curr. Opin. Cell Biol. 2018, 51, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, M.Y.; Sun, M.; Jiang, L.Y.; Zhao, X.T.; Fang, X.X.; Lam, S.M.; Shui, G.H.; Luo, J.; Shi, X.J.; et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J. Lipid Res. 2019, 60, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N.; Sheldon, K.L.; Dehart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodríguez Martínez, M.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating compound mechanismof action by network perturbation analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar] [PubMed]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sui, S.; Wang, L.; Li, H.; Zhang, L.; Xu, S.; Zheng, X. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell. Physiol. 2020, 235, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, Growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits systemxc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Jiang, X.; Dong, Z.; Hu, S.; Xiao, M. Low-concentration PTX And RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag. Res. 2019, 11, 9783–9792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobler, L.; Zhang, H.; Suri, P.; Park, C.; Timmerman, L.A. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget 2018, 9, 32280–32297. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol. Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef] [Green Version]
- Beyoğlu, D.; Idle, J.R. Metabolic Rewiring and the Characterization of Oncometabolites. Cancers 2021, 13, 2900. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iessi, E.; Vona, R.; Cittadini, C.; Matarrese, P. Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path. Biomedicines 2021, 9, 1942. https://doi.org/10.3390/biomedicines9121942
Iessi E, Vona R, Cittadini C, Matarrese P. Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path. Biomedicines. 2021; 9(12):1942. https://doi.org/10.3390/biomedicines9121942
Chicago/Turabian StyleIessi, Elisabetta, Rosa Vona, Camilla Cittadini, and Paola Matarrese. 2021. "Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path" Biomedicines 9, no. 12: 1942. https://doi.org/10.3390/biomedicines9121942
APA StyleIessi, E., Vona, R., Cittadini, C., & Matarrese, P. (2021). Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path. Biomedicines, 9(12), 1942. https://doi.org/10.3390/biomedicines9121942