
Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Aberrant TGF-β Signalling Underlies FOP
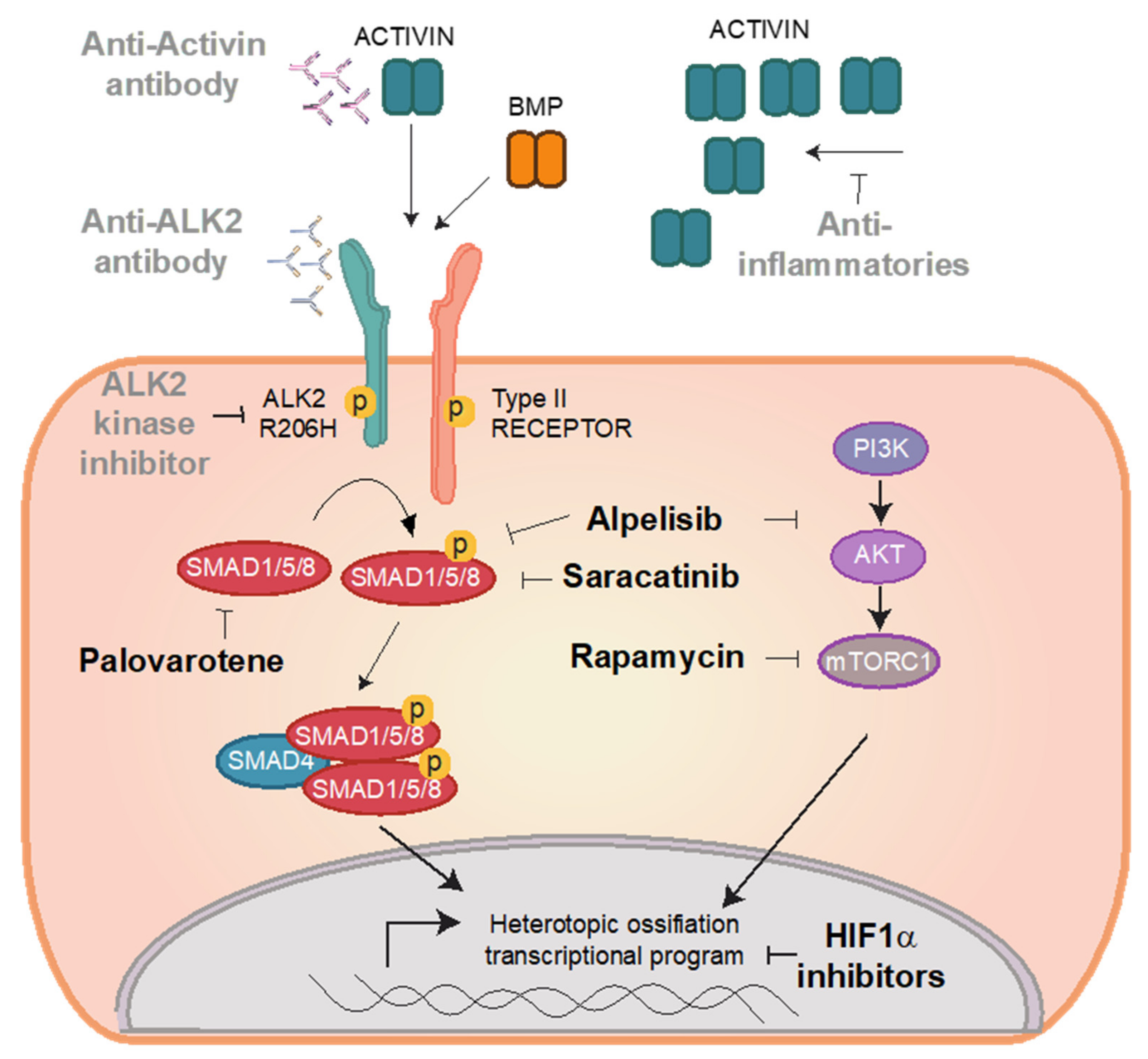
ALK2 Signalling in FOP
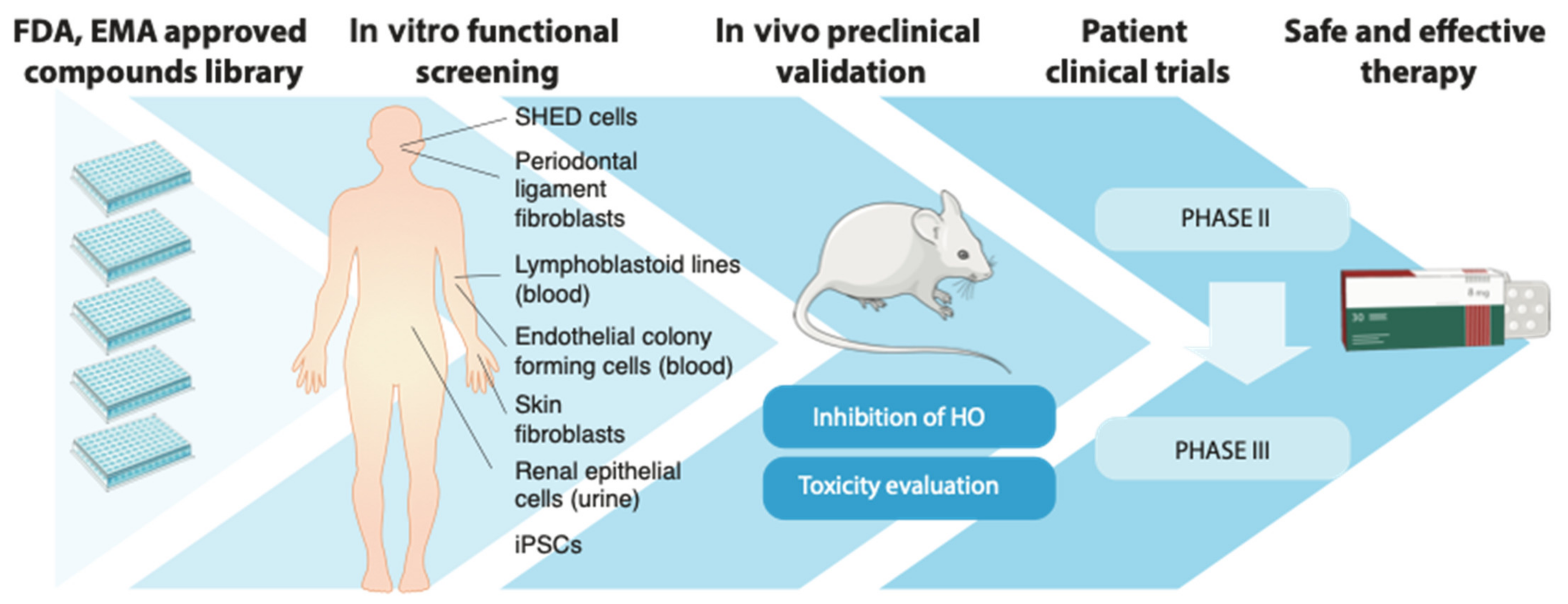
3. In Vitro Research Platforms Resembling FOP
4. Repurposed Drugs for FOP
4.1. Drug Repurposing versus De Novo Drug Development
4.2. Preclinical Candidates
4.3. Saracatinib
4.4. Rapamycin
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthrom, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Zasloff, M.A.; Kitterman, J.A.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J. Bone Jt. Surg. Am. 2010, 92, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Severino, M.; Bertamino, M.; Tortora, D.; Morana, G.; Uccella, S.; Bocciardi, R.; Ravazzolo, R.; Rossi, A.; Di Rocco, M. Novel asymptomatic CNS findings in patients with ACVR1/ALK2 mutations causing fibrodysplasia ossificans progressiva. J. Med. Genet. 2016, 53, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Kitterman, J.A.; Strober, J.B.; Kan, L.; Rocke, D.M.; Cali, A.; Peeper, J.; Snow, J.; Delai, P.L.; Morhart, R.; Pignolo, R.J.; et al. Neurological symptoms in individuals with fibrodysplasia ossificans progressiva. J. Neurol 2012, 259, 2636–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, L.; Kitterman, J.A.; Procissi, D.; Chakkalakal, S.; Peng, C.Y.; McGuire, T.L.; Goldsby, R.E.; Pignolo, R.J.; Shore, E.M.; Kaplan, F.S.; et al. CNS demyelination in fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2644–2655. [Google Scholar] [CrossRef] [Green Version]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Manganaro, A.; Calabro, M.P.; Salpietro, C.; Gitto, E. Fibrodysplasia ossificans progressiva in a newborn with cardiac involvement. Pediatr. Int. 2015, 57, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Kou, S.; De Cunto, C.; Baujat, G.; Wentworth, K.L.; Grogan, D.R.; Brown, M.A.; Di Rocco, M.; Keen, R.; Al Mukaddam, M.; le Quan Sang, K.H.; et al. Patients with ACVR1(R206H) mutations have an increased prevalence of cardiac conduction abnormalities on electrocardiogram in a natural history study of Fibrodysplasia Ossificans Progressiva. Orphanet. J. Rare Dis. 2020, 15, 193. [Google Scholar] [CrossRef]
- Ware, A.D.; Brewer, N.; Meyers, C.; Morris, C.; McCarthy, E.; Shore, E.M.; James, A.W. Differential Vascularity in Genetic and Nonhereditary Heterotopic Ossification. Int. J. Surg. Pathol. 2019, 27, 859–867. [Google Scholar] [CrossRef]
- Wentworth, K.L.; Bigay, K.; Chan, T.V.; Ho, J.P.; Morales, B.M.; Connor, J.; Brooks, E.; Shahriar Salamat, M.; Sanchez, H.C.; Wool, G.; et al. Clinical-pathological correlations in three patients with fibrodysplasia ossificans progressiva. Bone 2018, 109, 104–110. [Google Scholar] [CrossRef]
- El-Labban, N.G.; Hopper, C.; Barber, P. Ultrastructural finding of vascular degeneration in fibrodysplasia ossificans progressiva (FOP). J. Oral. Pathol. Med. 1995, 24, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Benderitter, P.; Orlova, V.; van Wijhe, M.; Garcia de Vinuesa, A.; Kerr, G.; Caradec, J.; Lodder, K.; de Boer, H.C.; et al. Development of Macrocycle Kinase Inhibitors for ALK2 Using Fibrodysplasia Ossificans Progressiva-Derived Endothelial Cells. JBMR Plus 2019, 3, e10230. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Heterotopic bone induction via BMP signaling: Potential therapeutic targets for fibrodysplasia ossificans progressiva. Bone 2018, 109, 241–250. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Glaser, D.L.; Collins, F.; Connor, M.; Kitterman, J.; Sillence, D.; Zackai, E.; Ravitsky, V.; Zasloff, M.; et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008, 121, e1295–e1300. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Lounev, V.Y.; Wang, H.; Pignolo, R.J.; Shore, E.M. Fibrodysplasia ossificans progressiva: A blueprint for metamorphosis. Ann. N. Y. Acad. Sci. 2011, 1237, 5–10. [Google Scholar] [CrossRef]
- Sell, S.; Willms, R.; Jany, R.; Esenwein, S.; Gaissmaier, C.; Martini, F.; Bruhn, G.; Burkhardsmaier, F.; Bamberg, M.; Kusswetter, W. The suppression of heterotopic ossifications: Radiation versus NSAID therapy—A prospective study. J. Arthroplast. 1998, 13, 854–859. [Google Scholar] [CrossRef]
- Karunakar, M.A.; Sen, A.; Bosse, M.J.; Sims, S.H.; Goulet, J.A.; Kellam, J.F. Indometacin as prophylaxis for heterotopic ossification after the operative treatment of fractures of the acetabulum. J. Bone Jt. Surg. Br. 2006, 88, 1613–1617. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Mechanistic insight into contextual TGF-beta signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb Perspect. Biol. 2016, 8, a021899-021828. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-β Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a022269-022242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci Signal. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Piek, E.; Afrakhte, M.; Sampath, K.; van Zoelen, E.J.; Heldin, C.H.; ten Dijke, P. Functional antagonism between activin and osteogenic protein-1 in human embryonal carcinoma cells. J. Cell. Physiol. 1999, 180, 141–149. [Google Scholar] [CrossRef]
- Olsen, O.E.; Sankar, M.; Elsaadi, S.; Hella, H.; Buene, G.; Darvekar, S.R.; Misund, K.; Katagiri, T.; Knaus, P.; Holien, T. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. J. Cell Sci. 2018, 131, jcs.213512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, O.E.; Hella, H.; Elsaadi, S.; Jacobi, C.; Martinez-Hackert, E.; Holien, T. Activins as Dual Specificity TGF-beta Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules 2020, 10, 519. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Chen, Y.G. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell. Physiol. 2017, 233, 4077–4099. [Google Scholar] [CrossRef]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landstrom, M. TGF-beta promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85alpha. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, A.; von Bulow, V.; Hamidi, R.; Winssinger, N.; Barluenga, S.; Heldin, C.H.; Landstrom, M. Polyubiquitination of transforming growth factor beta (TGFbeta)-associated kinase 1 mediates nuclear factor-kappaB activation in response to different inflammatory stimuli. J. Biol. Chem. 2012, 287, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, N.; Sorrentino, A.; Heldin, C.H.; Landstrom, M. TGF-beta uses the E3-ligase TRAF6 to turn on the kinase TAK1 to kill prostate cancer cells. Future Oncol. 2009, 5, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Landstrom, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Doody, J.; Massague, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Janda, E.; Pierreux, C.E.; Rytomaa, M.; Schulze, A.; McMahon, M.; Hill, C.S.; Beug, H.; Downward, J. Raf induces TGFbeta production while blocking its apoptotic but not invasive responses: A mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000, 14, 2610–2622. [Google Scholar] [CrossRef] [Green Version]
- Javelaud, D.; Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: Implications for carcinogenesis. Oncogene 2005, 24, 5742–5750. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Vasilaki, E.; Voytyuk, O.; Bai, Y.; Morikawa, M.; Moustakas, A.; Miyazono, K.; Heldin, C.H.; Ten Dijke, P.; van Dam, H. TGFbeta and EGF signaling orchestrates the AP-1- and p63 transcriptional regulation of breast cancer invasiveness. Oncogene 2020, 39, 4436–4449. [Google Scholar] [CrossRef]
- Sundqvist, A.; Morikawa, M.; Ren, J.; Vasilaki, E.; Kawasaki, N.; Kobayashi, M.; Koinuma, D.; Aburatani, H.; Miyazono, K.; Heldin, C.H.; et al. JUNB governs a feed-forward network of TGFbeta signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018, 46, 1180–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrecchia, F.; Tacheau, C.; Wagner, E.F.; Mauviel, A. A central role for the JNK pathway in mediating the antagonistic activity of pro-inflammatory cytokines against transforming growth factor-beta-driven SMAD3/4-specific gene expression. J. Biol. Chem. 2003, 278, 1585–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Pignolo, R.J.; Shore, E.M. The FOP metamorphogene encodes a novel type I receptor that dysregulates BMP signaling. Cytokine Growth Factor Rev. 2009, 20, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, J.; Xu, M.; Shore, E.M. Variable signaling activity by FOP ACVR1 mutations. Bone 2018, 109, 232–240. [Google Scholar] [CrossRef]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef] [Green Version]
- Machiya, A.; Tsukamoto, S.; Ohte, S.; Kuratani, M.; Fujimoto, M.; Kumagai, K.; Osawa, K.; Suda, N.; Bullock, A.N.; Katagiri, T. Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 2018, 111, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hino, k.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, J.; Deichsel, A.; Stange, K.; Ast, C.; Bocciardi, R.; Ravazzolo, R.; di Rocco, M.; Ferrari, P.; Landi, A.; Kaplan, F.S.; et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum. Mol. Genet. 2014, 23, 5364–5377. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Tajer, B.; Shore, E.M.; Mullins, M.C. Fibrodysplasia ossificans progressiva mutant ACVR1 signals by multiple modalities in the developing zebrafish. Elife 2020, 9, e53761. [Google Scholar] [CrossRef]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323 e312. [Google Scholar] [CrossRef]
- Groppe, J.C.; Wu, J.; Shore, E.M.; Kaplan, F.S. In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 2011, 194, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [Green Version]
- van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef]
- Chen, Y.G.; Liu, F.; Massague, J. Mechanism of TGFbeta receptor inhibition by FKBP12. Embo J. 1997, 16, 3866–3876. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Attisano, L.; Carcamo, J.; Ventura, F.; Weis, F.M.; Massague, J.; Wrana, J.L. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75, 671–680. [Google Scholar] [CrossRef]
- Ebner, R.; Chen, R.H.; Shum, L.; Lawler, S.; Zioncheck, T.F.; Lee, A.; Lopez, A.R.; Derynck, R. Cloning of a type I TGF-beta receptor and its effect on TGF-beta binding to the type II receptor. Science 1993, 260, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Macias-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, K.; Souchelnytskyi, S.; Itoh, S.; Nakao, A.; Sampath, K.; Heldin, C.H.; ten Dijke, P. Intracellular signaling of osteogenic protein-1 through Smad5 activation. J. Cell. Physiol. 1998, 177, 355–363. [Google Scholar] [CrossRef]
- ten Dijke, P.; Yamashita, H.; Sampath, T.K.; Reddi, A.H.; Estevez, M.; Riddle, D.L.; Ichijo, H.; Heldin, C.H.; Miyazono, K. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J. Biol. Chem. 1994, 269, 16985–16988. [Google Scholar] [CrossRef]
- Gipson, G.R.; Goebel, E.J.; Hart, K.N.; Kappes, E.C.; Kattamuri, C.; McCoy, J.C.; Thompson, T.B. Structural perspective of BMP ligands and signaling. Bone 2020, 140, 115549. [Google Scholar] [CrossRef]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. Elife 2020, 9, e54582. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Duffhues, G.; de Gorter, D.J.; Ten Dijke, P. Towards a cure for Fibrodysplasia ossificans progressiva. Ann. Transl. Med. 2016, 4, S28. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; Fotsis, T.; Dijke, P.T. Signal Transduction: Gain of Activin Turns Muscle into Bone. Curr. Biol. 2015, 25, R1136–R1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1(R206H) FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2019, 30, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, L.; Stange, K.; Deichsel, A.; Gossen, M.; Seemann, P. The Fibrodysplasia Ossificans Progressiva (FOP) mutation p.R206H in ACVR1 confers an altered ligand response. Cell Signal. 2017, 29, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Fiori, J.L.; Billings, P.C.; de la Pena, L.S.; Kaplan, F.S.; Shore, E.M. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2006, 21, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valer, J.A.; Sanchez-de-Diego, C.; Gamez, B.; Mishina, Y.; Rosa, J.L.; Ventura, F. Inhibition of phosphatidylinositol 3-kinase alpha (PI3Kalpha) prevents heterotopic ossification. Embo Mol. Med. 2019, 11, e10567. [Google Scholar] [CrossRef]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Eekhoff, E.M.W.; Botman, E.; Coen Netelenbos, J.; de Graaf, P.; Bravenboer, N.; Micha, D.; Pals, G.; de Vries, T.J.; Schoenmaker, T.; Hoebink, M.; et al. [18F]NaF PET/CT scan as an early marker of heterotopic ossification in fibrodysplasia ossificans progressiva. Bone 2018, 109, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Gannon, F.H.; Kaplan, F.S.; Olmsted, E.; Finkel, G.C.; Zasloff, M.A.; Shore, E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum. Pathol. 1997, 28, 339–343. [Google Scholar] [CrossRef]
- Virdi, A.S.; Shore, E.M.; Oreffo, R.O.; Li, M.; Connor, J.M.; Smith, R.; Kaplan, F.S.; Triffitt, J.T. Phenotypic and molecular heterogeneity in fibrodysplasia ossificans progressiva. Calcif. Tissue Int. 1999, 65, 250–255. [Google Scholar] [CrossRef]
- Olmsted, E.A.; Kaplan, F.S.; Shore, E.M. Bone morphogenetic protein-4 regulation in fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 2003, 331–343. [Google Scholar] [CrossRef]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef]
- Bagarova, J.; Vonner, A.J.; Armstrong, K.A.; Borgermann, J.; Lai, C.S.; Deng, D.Y.; Beppu, H.; Alfano, I.; Filippakopoulos, P.; Morrell, N.W.; et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell Biol 2013, 33, 2413–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.-A.; Kim, H.-J.; Woo, K.-M.; Baek, J.-H.; Kim, G.-S.; Choi, J.-Y.; Ryoo, H.-M. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J. Biol. Chem. 2010, 285, 22542–22553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culbert, A.L.; Chakkalakal, S.A.; Theosmy, E.G.; Brennan, T.A.; Kaplan, F.S.; Shore, E.M. Alk2 Regulates Early Chondrogenic Fate in Fibrodysplasia Ossificans Progressiva Heterotopic Endochondral Ossification. Stem Cells 2014, 32, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- de la Peña, L.S.; Billings, P.C.; Fiori, J.L.; Ahn, J.; Kaplan, F.S.; Shore, E.M. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J. Bone Miner. Res. 2005, 20, 1168–1176. [Google Scholar] [CrossRef]
- Del Zotto, G.; Antonini, F.; Azzari, I.; Ortolani, C.; Tripodi, G.; Giacopelli, F.; Cappato, S.; Moretta, L.; Ravazzolo, R.; Bocciardi, R. Peripheral blood mononuclear cell immunophenotyping in fibrodysplasia ossificans progressiva patients: Evidence for monocyte DNAM1 up-regulation. Cytom. B Clin. Cytom 2017, 94, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Micha, D.; Voermans, E.; Eekhoff, M.E.W.; van Essen, H.W.; Zandieh-Doulabi, B.; Netelenbos, C.; Rustemeyer, T.; Sistermans, E.A.; Pals, G.; Bravenboer, N. Inhibition of TGFβ signaling decreases osteogenic differentiation of fibrodysplasia ossificans progressiva fibroblasts in a novel in vitro model of the disease. Bone 2016, 84, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; DECKERS, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; Ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, T.J.; Schoenmaker, T.; Micha, D.; Hogervorst, J.; Bouskla, S.; Forouzanfar, T.; Pals, G.; Netelenbos, C.; Eekhoff, E.M.W.; Bravenboer, N. Periodontal ligament fibroblasts as a cell model to study osteogenesis and osteoclastogenesis in fibrodysplasia ossificans progressiva. Bone 2018, 109, 168–177. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Wouters, F.; Micha, D.; Forouzanfar, T.; Netelenbos, C.; Eekhoff, E.M.W.; Bravenboer, N.; de Vries, T.J. The effect of Activin-A on periodontal ligament fibroblasts-mediated osteoclast formation in healthy donors and in patients with fibrodysplasia ossificans progressiva. J. Cell Physiol 2019, 234, 10238–10247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 2013, 140, 2457–2461. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Nagata, N.; Kurokawa, H.; Yamanaka, S. iPS cells: A game changer for future medicine. Embo J. 2014, 33, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Hashizume, Y.; Yamada, Y.; Katayama, T.; Hohjoh, H.; Fusaki, N.; Nakashima, Y.; Furuya, H.; Haga, N.; Takami, Y.; et al. Pathogenic Mutation of ALK2 Inhibits Induced Pluripotent Stem Cell Reprogramming and Maintenance: Mechanisms of Reprogramming and Strategy for Drug Identification. Stem Cells 2012, 30, 2437–2449. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Mikkers, H.; de Jong, D.; Szuhai, K.; de Vries, T.J.; Freund, C.; Bravenboer, N.; van Es, R.J.J.; Netelenbos, J.C.; Goumans, M.J.; et al. Generation of Fibrodysplasia ossificans progressiva and control integration free iPSC lines from periodontal ligament fibroblasts. Stem Cell Res. 2019, 41, 101639. [Google Scholar] [CrossRef]
- Cai, J.; Orlova, V.V.; Cai, X.; Eekhoff, E.M.W.; Zhang, K.; Pei, D.; Pan, G.; Mummery, C.L.; Ten Dijke, P. Induced Pluripotent Stem Cells to Model Human Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2015, 5, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ikeya, M.; Hino, K.; Horigome, K.; Fukuta, M.; Watanabe, M.; Nagata, S.; Yamamoto, T.; Otsuka, T.; Toguchida, J. New Protocol to Optimize iPS Cells for Genome Analysis of Fibrodysplasia Ossificans Progressiva. Stem Cells 2015, 33, 1730–1742. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet. J. Rare Dis 2013, 8, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, T.; Shibata, M.; Nishio, M.; Nagata, S.; Alev, C.; Sakurai, H.; Toguchida, J.; Ikeya, M. Modeling human somite development and fibrodysplasia ossificans progressiva with induced pluripotent stem cells. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barruet, E.; Morales, B.M.; Lwin, W.; White, M.P.; Theodoris, C.V.; Kim, H.; Urrutia, A.; Wong, S.A.; Srivastava, D.; Hsiao, E.C. The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem Cell Res. 2016, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Lounev, V.Y.; Ramachandran, R.; Wosczyna, M.N.; Yamamoto, M.; Maidment, A.D.; Shore, E.M.; Glaser, D.L.; Goldhamer, D.J.; Kaplan, F.S. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Jt. Surg Am. 2009, 91, 652–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Q.; Sun, M.H.; Cheng, H.; Peng, Y.; Montag, A.G.; Deyrup, A.T.; Jiang, W.; Luu, H.H.; Luo, J.; Szatkowski, J.P.; et al. Characterization of the distinct orthotopic bone-forming activity of 14 BMPs using recombinant adenovirus-mediated gene delivery. Gene Ther. 2004, 11, 1312–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.A.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khairnar, K.J.; Jadhav, A.G. Drug Repurposing (DR): An Emerging Approach in Drug Discovery; IntechOpen: London, UK, 2020. [Google Scholar]
- Houslay, M.D. Melanoma, Viagra, and PDE5 Inhibitors: Proliferation and Metastasis. Trends Cancer 2016, 2, 163–165. [Google Scholar] [CrossRef] [Green Version]
- Badria, F.A.; Fayed, H.A.; Ibraheem, A.K.; State, A.F.; Mazyed, E.A. Formulation of Sodium Valproate Nanospanlastics as a Promising Approach for Drug Repurposing in the Treatment of Androgenic Alopecia. Pharmaceutics 2020, 12, 866. [Google Scholar] [CrossRef]
- Gao, S.; Wang, S.; Fan, R.; Hu, J. Recent advances in the molecular mechanism of thalidomide teratogenicity. Biomed. Pharm. 2020, 127, 110114. [Google Scholar] [CrossRef]
- Brown, P.M.; Isaacs, J.D. Rheumatoid arthritis: An evolutionary force in biologics. Curr Pharm Des. 2015, 21, 2170–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessetto, Z.Y.; Weir, S.J.; Sethi, G.; Broward, M.A.; Godwin, A.K. Drug repurposing for gastrointestinal stromal tumor. Mol. Cancer 2013, 12, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Simonis, A.; Theobald, S.J.; Fatkenheuer, G.; Rybniker, J.; Malin, J.J. A comparative analysis of remdesivir and other repurposed antivirals against SARS-CoV-2. Embo Mol. Med. 2020, 13, e13105. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Mateus, A.; Maatta, T.A.; Savitski, M.M. Thermal proteome profiling: Unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2016, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Joice, M.; Vasileiadis, G.I.; Amanatullah, D.F. Non-steroidal anti-inflammatory drugs for heterotopic ossification prophylaxis after total hip arthroplasty: A systematic review and meta-analysis. Bone Jt. J. 2018, 100-B, 915–922. [Google Scholar] [CrossRef]
- Brunnekreef, J.J.; Hoogervorst, P.; Ploegmakers, M.J.; Rijnen, W.H.; Schreurs, B.W. Is etoricoxib effective in preventing heterotopic ossification after primary total hip arthroplasty? Int. Orthop. 2013, 37, 583–587. [Google Scholar] [CrossRef]
- Lavernia, C.J.; Contreras, J.S.; Villa, J.M.; Rossi, M.D. Celecoxib and heterotopic bone formation after total hip arthroplasty. J. Arthroplast. 2014, 29, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, L.; Zhang, S.; Yu, B.; Liu, F.; Cui, Z.; Jin, D.; Bai, X. Celecoxib inhibits the heterotopic ossification in the rat model with Achilles tenotomy. Eur. J. Orthop. Surg. Traumatol. 2013, 23, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Jani, P.H.; Gibson, M.P.; Liu, C.; Zhang, H.; Wang, X.; Lu, Y.; Qin, C. Transgenic expression of Dspp partially rescued the long bone defects of Dmp1-null mice. Matrix Biol. 2016, 52–54, 95–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Liu, H.; Li, J.; Zhou, Y. Roles of hypoxia during the chondrogenic differentiation of mesenchymal stem cells. Curr Stem Cell Res. 2014, 9, 141–147. [Google Scholar] [CrossRef]
- Wang, H.; Lindborg, C.; Lounev, V.; Kim, J.H.; McCarrick-Walmsley, R.; Xu, M.; Mangiavini, L.; Groppe, J.C.; Shore, E.M.; Schipani, E.; et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J. Bone Miner. Res. 2016, 31, 1652–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Loder, S.; Brownley, C.; Cholok, D.; Mangiavini, L.; Li, J.; Breuler, C.; Sung, H.H.; Li, S.; Ranganathan, K.; et al. Inhibition of Hif1alpha prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl Acad Sci USA 2016, 113, E338–E347. [Google Scholar] [CrossRef] [Green Version]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Andolina, J.R.; Adamson, P.C.; Teachey, D.T.; Finklestein, J.Z.; Ebb, D.H.; Whitehead, B.; Jacobs, B.; Siegel, D.M.; Keen, R.; et al. Early clinical observations on the use of imatinib mesylate in FOP: A report of seven cases. Bone 2018, 109, 276–280. [Google Scholar] [CrossRef]
- Yamamoto, R.; Matsushita, M.; Kitoh, H.; Masuda, A.; Ito, M.; Katagiri, T.; Kawai, T.; Ishiguro, N.; Ohno, K. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J. Bone Miner. Metab. 2013, 31, 26–33. [Google Scholar] [CrossRef]
- Kitoh, H.; Achiwa, M.; Kaneko, H.; Mishima, K.; Matsushita, M.; Kadono, I.; Horowitz, J.D.; Sallustio, B.C.; Ohno, K.; Ishiguro, N. Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: An open-labeled clinical trial. Orphanet. J. Rare Dis. 2013, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Hind, M.; Stinchcombe, S. Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema. Curr Opin. Investig. Drugs 2009, 10, 1243–1250. [Google Scholar] [PubMed]
- Stolk, J.; Cooper, B.G.; Stoel, B.; Rames, A.; Rutman, O.; Soliman, S.; Stockley, R. Retinoid treatment of Emphysema in Patients on the Alpha-1 International Registry. The REPAIR study: Study design, methodology and quality control of study assessments. Adv. Respir. Dis 2010, 4, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Stolk, J.; Stockley, R.A.; Stoel, B.C.; Cooper, B.G.; Piitulainen, E.; Seersholm, N.; Chapman, K.R.; Burdon, J.G.; Decramer, M.; Abboud, R.T.; et al. Randomised controlled trial for emphysema with a selective agonist of the gamma-type retinoic acid receptor. Eur. Respir. J. 2012, 40, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, F.S.; Shore, E.M. Derailing heterotopic ossification and RARing to go. Nat. Med. 2011, 17, 420–421. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Mali, R.S.; Munugalavadla, V.; Krishnan, S.; Ramdas, B.; Sims, E.; Martin, H.; Ghosh, J.; Li, S.; Chan, R.J.; et al. The PI3K pathway drives the maturation of mast cells via microphthalmia transcription factor. Blood 2011, 118, 3459–3469. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 558, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Andre, F.; Yamashita, T.; Cerda, H.; Toledano, I.; Stemmer, S.M.; Jurado, J.C.; Juric, D.; Mayer, I.; Ciruelos, E.M.; et al. Time course and management of key adverse events during the randomized phase III SOLAR-1 study of PI3K inhibitor alpelisib plus fulvestrant in patients with HR-positive advanced breast cancer. Ann. Oncol. 2020, 31, 1001–1010. [Google Scholar] [CrossRef]
- Gamez, B.; Rodriguez-Carballo, E.; Graupera, M.; Rosa, J.L.; Ventura, F. Class I PI-3-Kinase Signaling Is Critical for Bone Formation Through Regulation of SMAD1 Activity in Osteoblasts. J. Bone Miner. Res. 2016, 31, 1617–1630. [Google Scholar] [CrossRef] [Green Version]
- Rokutanda, S.; Fujita, T.; Kanatani, N.; Yoshida, C.A.; Komori, H.; Liu, W.; Mizuno, A.; Komori, T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009, 328, 78–93. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Flanagan, J.U.; Kolekar, S.; Buchanan, C.; Kendall, J.D.; Lee, W.J.; Rewcastle, G.W.; Denny, W.A.; Singh, R.; Dickson, J.; et al. A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem. J. 2011, 438, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Lim, H.W.; Lee, S.H.; Han, H.J. Smad, PI3K/Akt, and Wnt-dependent signaling pathways are involved in BMP-4-induced ESC self-renewal. Stem Cells 2009, 27, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.T.; Dey, D.; Sanders, E.M.; Seavey, J.G.; Tomasino, A.M.; Moss, K.; Wheatley, B.; Cholok, D.; Loder, S.; Li, J.; et al. Inhibition of Mammalian Target of Rapamycin Signaling with Rapamycin Prevents Trauma-Induced Heterotopic Ossification. Am. J. Pathol. 2017, 187, 2536–2545. [Google Scholar] [CrossRef] [Green Version]
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; Lambert-van der Brempt, C.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef]
- Hannon, R.A.; Clack, G.; Rimmer, M.; Swaisland, A.; Lockton, J.A.; Finkelman, R.D.; Eastell, R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: A randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J. Bone Miner. Res. 2010, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res. 2015, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- McNeish, I.A.; Ledermann, J.A.; Webber, L.; James, L.; Kaye, S.B.; Hall, M.; Hall, G.; Clamp, A.; Earl, H.; Banerjee, S.; et al. A randomised, placebo-controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum-resistant ovarian, fallopian tube or primary peritoneal cancerdagger. Ann. Oncol. 2014, 25, 1988–1995. [Google Scholar] [CrossRef]
- Laurie, S.A.; Goss, G.D.; Shepherd, F.A.; Reaume, M.N.; Nicholas, G.; Philip, L.; Wang, L.; Schwock, J.; Hirsh, V.; Oza, A.; et al. A phase II trial of saracatinib, an inhibitor of src kinases, in previously-treated advanced non-small-cell lung cancer: The princess margaret hospital phase II consortium. Clin. Lung Cancer 2014, 15, 52–57. [Google Scholar] [CrossRef]
- Posadas, E.M.; Ahmed, R.S.; Karrison, T.; Szmulewitz, R.Z.; O’Donnell, P.H.; Wade, J.L., 3rd; Shen, J.; Gururajan, M.; Sievert, M.; Stadler, W.M. Saracatinib as a metastasis inhibitor in metastatic castration-resistant prostate cancer: A University of Chicago Phase 2 Consortium and DOD/PCF Prostate Cancer Clinical Trials Consortium Study. Prostate 2016, 76, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.M.; Kopetz, S.; Morris, J.; Parikh, N.; Qiao, W.; Overman, M.J.; Fogelman, D.; Shureiqi, I.; Jacobs, C.; Malik, Z.; et al. Phase II study of saracatinib (AZD0530) in patients with previously treated metastatic colorectal cancer. Invest. New Drugs 2015, 33, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, W.A.; Nallapareddy, S.; Arcaroli, J.; Tan, A.; Foster, N.R.; Wright, J.J.; Picus, J.; Goh, B.C.; Hidalgo, M.; Erlichman, C. A phase II trial of saracatinib (AZD0530), an oral Src inhibitor, in previously treated metastatic pancreatic cancer. J. Clin. Oncol. 2010, 28, e14515. [Google Scholar] [CrossRef]
- Frail, D.E.; Brady, M.; Escott, K.J.; Holt, A.; Sanganee, H.J.; Pangalos, M.N.; Watkins, C.; Wegner, C.D. Pioneering government-sponsored drug repositioning collaborations: Progress and learning. Nat. Rev. Drug Discov 2015, 14, 833–841. [Google Scholar] [CrossRef]
- Tyryshkin, A.; Bhattacharya, A.; Eissa, N.T. SRC kinase is a novel therapeutic target in lymphangioleiomyomatosis. Cancer Res. 2014, 74, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dyck, C.H.; Nygaard, H.B.; Chen, K.; Donohue, M.C.; Raman, R.; Rissman, R.A.; Brewer, J.B.; Koeppe, R.A.; Chow, T.W.; Rafii, M.S.; et al. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol 2019, 76, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.; Bagarova, J.; Kerr, G.; Xia, D.-D.; Place, E.S.; Dey, D.; Shen, Y.; Bocobo, G.A.; Mohedas, A.H.; Huang, X.; et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharm. Sci 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, T.; Scott, G.; Komatsu, Y.; Araya, R.; Kawano, M.; Ray, M.K.; Yamada, M.; Mishina, Y. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 2006, 44, 159–167. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Wentworth, K.L.; Masharani, U.; Hsiao, E.C. Therapeutic advances for blocking heterotopic ossification in fibrodysplasia ossificans progressiva. Br. J. Clin. Pharm. 2019, 85, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Zeitlin, L.; Dunn, S.P.; Benor, S.; Hagin, D.; Al Mukaddam, M.; Pignolo, R.J. Acute and chronic rapamycin use in patients with Fibrodysplasia Ossificans Progressiva: A report of two cases. Bone 2018, 109, 281–284. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Nicholas, S.-A.E.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. Elife 2018, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Goldhamer, D.J.; Lees-Shepard, J.B. Response to comment on Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. Elife 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Ipsen. Ipsen Provides Update on Palovarotene Clinical Programs. Available online: https://www.ipsen.com/websites/Ipsen_Online/wp-content/uploads/2020/03/25224057/Ipsen-Press-Release-Palovarotene-Update-ENGLISH.pdf (accessed on 26 March 2020).
- Regeneron. Regeneron Provides Update on the Garetosmab Phase 2 LUMINA-1 Trial in Fibrodysplasia Ossificans progressiva (FOP). Available online: https://investor.regeneron.com/static-files/1a038387-67e6-4446-91fa-813b0225235e (accessed on 30 October 2020).
- Claudia Ordonez, S.B.; Barger, R.; Serino, T.; Tseng, C.C.; Rovaldi, C.; Lachey, J.; Seehra, J. Administration of KER-047, a Novel ALK2 Inhibitor, Elicited Robust and Sustained Increases in Serum Iron in Healthy Participants. Blood 2020, 136, 13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Repurposing in Rare Diseases | |
|---|---|
| Benefits | Disadvantages |
| Lower investment required | Patent already filed, might require negotiation |
| Possibility of synergistic effect by targeting different pathways simultaneously | Drug candidates may not be clean and have additional unwanted targets |
| Compound libraries commercially available | |
| Drug-like properties already characterized | |
| Lower number of patients in trials | |
| Shorter development time | |
| Limited risk of failure | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventura, F.; Williams, E.; Ikeya, M.; Bullock, A.N.; ten Dijke, P.; Goumans, M.-J.; Sanchez-Duffhues, G. Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 213. https://doi.org/10.3390/biomedicines9020213
Ventura F, Williams E, Ikeya M, Bullock AN, ten Dijke P, Goumans M-J, Sanchez-Duffhues G. Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva. Biomedicines. 2021; 9(2):213. https://doi.org/10.3390/biomedicines9020213
Chicago/Turabian StyleVentura, Francesc, Eleanor Williams, Makoto Ikeya, Alex N. Bullock, Peter ten Dijke, Marie-José Goumans, and Gonzalo Sanchez-Duffhues. 2021. "Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva" Biomedicines 9, no. 2: 213. https://doi.org/10.3390/biomedicines9020213
APA StyleVentura, F., Williams, E., Ikeya, M., Bullock, A. N., ten Dijke, P., Goumans, M. -J., & Sanchez-Duffhues, G. (2021). Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva. Biomedicines, 9(2), 213. https://doi.org/10.3390/biomedicines9020213