
Metabolic Diseases and Down Syndrome: How Are They Linked Together?


 ,
,  , ,
, , 
{kind=link}
Abstract
:1. Introduction
1.1. Down Syndrome
1.1.1. Genotype and Phenotypes
1.1.2. DS Comorbidities
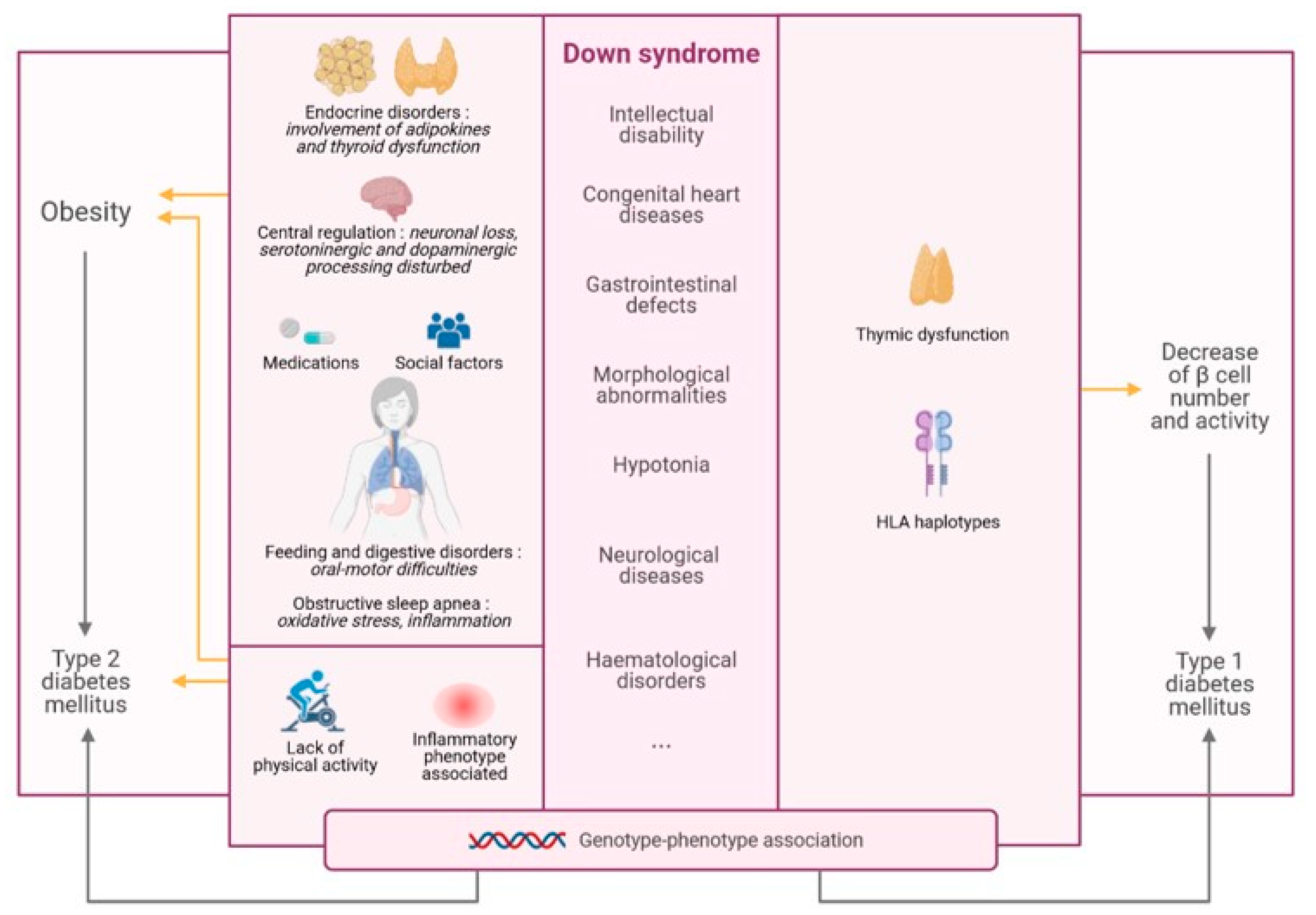
1.2. Statement of Hypothesis
2. DS and Obesity
2.1. Increased Prevalence of Obesity in DS
2.2. Physical Activity in DS
2.3. Endocrine Disorders in DS
2.3.1. Involvement of White Adipose Tissue—Adipokines
2.3.2. Thyroid Dysfunction in DS
2.4. Feeding and Digestive Disorders
2.5. Inflammation
2.6. Obstructive Sleep Apnea
2.7. Social Factors
2.8. Medications
2.9. Central Regulation of Food Intake
3. DS and Diabetes Mellitus
3.1. T1DM and DS
3.1.1. T1DM in DS: A Link with Immune System Defects
3.1.2. Genotype–Phenotype Association and T1DM-DS
3.2. T2DM and DS
3.2.1. T2DM in DS: A Link with Obesity
3.2.2. Genotype–Phenotype Association and T2DM-DS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AgRP | agouti-related protein |
| APP | Amyloid Precursor Protein |
| ARC | arcuate nucleus |
| AIRE | autoimmune regulator |
| BMI | body mass index |
| CART | cocaine- and amphetamine-related transcript |
| DMN | dorsomedial nucleus |
| DS | Down syndrome |
| DSCR | Down Syndrome Critical Region |
| DEXA | dual energy X-ray absorptiometry |
| DYRK1A | Dual specificity tyrosine-phosphorylation-regulated kinase 1 |
| GERD | gastroesophageal reflux disease |
| GSK3 β | Glycogen synthase kinase 3 beta |
| HDL | high density lipoprotein |
| HLA | Haplotype Leukocyte Antigen |
| HSA21 | human chromosome 21 |
| ID | intellectual disability |
| IL- | interleukin- |
| IL-1RA | interleukin 1 receptor antagonist |
| IFN-γ | interferon gamma |
| KO | knock-out |
| LDL | low density lipoprotein |
| LHA | lateral hypothalamic area |
| mTEC | medullary thymic epithelial cells |
| NAc | nucleus accumbens |
| NFAT | nuclear factor of activated T cells |
| NPY | neuropeptide Y |
| OSA | Obstructive sleep apnea |
| PBF | percentage of body fat |
| POMC | proopiomelanocortin |
| PVN | paraventricular nucleus |
| RCAN1 | Regulatory of Calcineurin 1 |
| SNV | single nucleotide variation |
| SOD1 | Superoxide dismutase 1 |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| T3 | 3,5,3′-triiodothyronine |
| T4 | Thyroxine |
| TH | thyroid hormone |
| TNF-α | Tumor necrosis factor alpha |
| TRH | Thyrotropin releasing hormone |
| TSH | Thyroid stimulating hormone |
| VMN | ventromedial hypothalamic nucleus |
| VTA | ventral tegmental area |
References
- Lakovschek, I.C.; Streubel, B.; Ulm, B. Natural outcome of trisomy 13, trisomy 18, and triploidy after prenatal diagnosis. Am. J. Med. Genet. Part A 2011, 155, 2626–2633. [Google Scholar] [CrossRef]
- Epstein, C.J. Developmental genetics. Cell. Mol. Life Sci. 1986, 42, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Al-Alaiyan, S.; Al-Omran, H.; Kattan, H.; Sakati, N.; Nyhan, W.L. Down Syndrome and Recurrent Abortions Resulting from Robertsonian Translocation 21q21q. Ann. Saudi Med. 1995, 15, 391–392. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.B.; Adelsberger, P.A.; Schinzel, A.A.; Hinkel, G.K.; Antonarakis, S.E. Down Syndrome Due to De Novo Robertsonian Translocation t(l 4q;21 q): DNA Polymorphism Analysis Suggests That the Origin of the Extra 21q Is Maternal. Am. J. Hum. Genet. 1991, 49, 529–536. [Google Scholar] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Prim. 2020, 6, 1–20. [Google Scholar] [CrossRef]
- Epstein, C.J. Down’s syndrome: Critical genes in a critical region. Nat. Cell Biol. 2006, 441, 582–583. [Google Scholar] [CrossRef]
- Yahya-Graison, E.A.; Aubert, J.; Dauphinot, L.; Rivals, I.; Prieur, M.; Golfier, G.; Rossier, J.; Personnaz, L.; Créau, N.; Bléhaut, H.; et al. Classification of Human Chromosome 21 Gene-Expression Variations in Down Syndrome: Impact on Disease Phenotypes. Am. J. Hum. Genet. 2007, 81, 475–491. [Google Scholar] [CrossRef] [Green Version]
- Delabar, J.-M.; Theophile, D.; Rahmani, Z.; Chettouh, Z.; Blouin, J.-L.; Prieur, M.; Noel, B.; Sinet, P.-M. Molecular Mapping of Twenty-Four Features of Down Syndrome on Chromosome 21. Eur. J. Hum. Genet. 1993, 1, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.E.; Richtsmeier, J.T.; Leszl, J.; Reeves, R.H. A Chromosome 21 Critical Region Does Not Cause Specific Down Syndrome Phenotypes. Science 2004, 306, 687–690. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and Down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Hervé, B.; Coussement, A.; Gilbert, T.; Dumont, F.; Jacques, S.; Cuisset, L.; Chicard, M.; Hizem, S.; Bourdoncle, P.; Letourneur, F.; et al. Aneuploidy: The impact of chromosome imbalance on nuclear organization and overall genome expression. Clin. Genet. 2016, 90, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.K.; Alford, K.A.; Tybulewicz, V.L.; Fisher, E.M. Down syndrome—Recent progress and future prospects. Hum. Mol. Genet. 2009, 18, R75–R83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofei, F. Obesity—A Preventable Disease. Ghana Med. J. 2005, 39, 98–101. [Google Scholar] [PubMed]
- Kissebah, A.H.; Krakower, G.R. Regional adiposity and morbidity. Physiol. Rev. 1994, 74, 761–811. [Google Scholar] [CrossRef]
- Klinerogerseva, R.; Eagle, T.F.; Sheetz, A.; Woodward, A.; Leibowitz, R.; Song, M.; Sylvester, R.; Corriveau, N.; Kline-Rogers, E.; Jiang, Q.; et al. The Relationship between Childhood Obesity, Low Socioeconomic Status, and Race/Ethnicity: Lessons from Massachusetts. Child. Obes. 2015, 11, 691–695. [Google Scholar] [CrossRef]
- Brantley, P.J.; Myers, V.H.; Roy, H.J. Environmental and lifestyle influences on obesity. J. La. State Med Soc. Off. Organ La. State Med. Soc. 2005, c, S19–S27. [Google Scholar]
- Herrera, B.M.; Lindgren, C.M. The Genetics of Obesity. Curr. Diabetes Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neel, J.V. Diabetes Mellitus: A “Thrifty” Genotype Rendered Detrimental by “Progress”? 1962. Bull. World Health Organ. 1999, 77, 694–703, discussion 692–693. [Google Scholar]
- Southam, L.; Soranzo, N.; Montgomery, S.B.; Frayling, T.M.; McCarthy, M.I.; Barroso, I.; Zeggini, E. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetol. 2009, 52, 1846–1851. [Google Scholar] [CrossRef] [Green Version]
- Speakman, J.R. Thrifty genes for obesity and the metabolic syndrome—Time to call off the search? Diabetes Vasc. Dis. Res. 2006, 3, 7–11. [Google Scholar] [CrossRef]
- Dubern, B.; Clement, K. Leptin and leptin receptor-related monogenic obesity. Biochimie 2012, 94, 2111–2115. [Google Scholar] [CrossRef]
- Ozsu, E.; Ceylaner, S.; Onay, H. Early-onset severe obesity due to complete deletion of the leptin gene in a boy. J. Pediatr. Endocrinol. Metab. 2017, 30, 1227–1230. [Google Scholar] [CrossRef]
- Shabana; Hasnain, S. The p. N103K mutation of leptin (LEP) gene and severe early onset obesity in Pakistan. Biol. Res. 2016, 49, 23. [Google Scholar] [CrossRef] [Green Version]
- Thaker, V.V. Genetic and epigenetic causes of obesity. Adolesc. Med. State Art Rev. 2017, 28, 379–405. [Google Scholar] [PubMed]
- Herrera, B.M.; Keildson, S.; Lindgren, C.M. Genetics and epigenetics of obesity. Maturitas 2011, 69, 41–49. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Gunnarsson, R.; Björkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Geisler, F.; Oikonen, V.; Mäki, M.; Takala, T.; Solin, O.; Ferrannini, E.; Knuuti, J.; Nuutila, P. 18F-FDG PET Study Insulin Stimulates Liver Glucose Uptake in Humans: An 18F-FDG PET Study. J. Nucl. Med. 2003, 44, 682–689. [Google Scholar] [PubMed]
- Virtanen, K.A.; Lönnroth, P.; Parkkola, R.; Peltoniemi, P.; Asola, M.; Viljanen, T.; Tolvanen, T.; Knuuti, J.; Rönnemaa, T.; Huupponen, R.; et al. Glucose Uptake and Perfusion in Subcutaneous and Visceral Adipose Tissue during Insulin Stimulation in Nonobese and Obese Humans. J. Clin. Endocrinol. Metab. 2002, 87, 3902–3910. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of Diabetes and Diabetes-Related Complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, E.; Aggarwal, A. Insulin Therapy for Patients with Type 1 Diabetes. J. Assoc. Physicians India 2007, 55, 29. [Google Scholar]
- Gregersen, P.K. HLA class II polymorphism: Implications for genetic susceptibility to autoimmune disease. Lab. Investig. 1989, 61, 5–19. [Google Scholar] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.A.; Rizza, R.A.; Butler, P.C. -Cell Deficit and Increased -Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Man, C.D.; Basu, R.; Toffolo, G.; Cobelli, C.; Rizza, R.A. Effects of Type 2 Diabetes on Insulin Secretion, Insulin Action, Glucose Effectiveness, and Postprandial Glucose Metabolism. Diabetes Care 2009, 32, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of Insulin Resistance by Hyperin-sulinaemia in Man. Diabetologia 1985, 28, 70–75. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Wang, C.; Guan, Y.; Yang, J. Cytokines in the Progression of Pancreaticβ-Cell Dysfunction. Int. J. Endocrinol. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, A.J.; Walker, J.D.; Frier, B.M. Type 1 Diabetes Mellitus and Down’s Syndrome: Prevalence, Manage-ment and Diabetic Complications. Diabet. Med. 1998, 15, 160–163. [Google Scholar] [CrossRef]
- Farquhar, J. Early-onset diabetes in the general and the down’s syndrome population. Lancet 1969, 294, 323–324. [Google Scholar] [CrossRef]
- Bell, A.J.; Bhate, M.S. Prevalence of overweight and obesity in Down’s syndrome and other mentally handicapped adults living in the community. J. Intellect. Disabil. Res. 2008, 36, 359–364. [Google Scholar] [CrossRef]
- Hawli, Y.; Nasrallah, M.; Fuleihan, G.E.-H. Endocrine and musculoskeletal abnormalities in patients with Down syndrome. Nat. Rev. Endocrinol. 2009, 5, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Petri, H.; Ding, Y.; Wandel, C.; Khwaja, O.; Foskett, N. Morbidity and medication in a large population of individuals with Down syndrome compared to the general population. Dev. Med. Child Neurol. 2015, 58, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Prasher, V.P. Overweight and obesity amongst Down’s syndrome adults. J. Intellect. Disabil. Res. 1995, 39, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Havercamp, S.M.; Scandlin, D.; Roth, M. Health Disparities among Adults with Developmental Disabilities, Adults with other Disabilities, and Adults Not Reporting Disability in North Carolina. Public Health Rep. 2004, 119, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.; Ramsey, K.; Pinter, J. Trends in Obesity and Overweight in Oregon Children with Down Syndrome. Glob. Pediatr. Health 2019, 6, 2333794X19835640. [Google Scholar] [CrossRef]
- Mircher, C.; Briceño, L.G.; Toulas, J.; Conte, M.; Tanguy, M.-L.; Cieuta-Walti, C.; Rethore, M.-O.; Ravel, A. Growth curves for French people with Down syndrome from birth to 20 years of age. Am. J. Med Genet. Part A 2018, 176, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- González-Agüero, A.; Ara, I.; Moreno, L.A.; Vicente-Rodríguez, G.; Casajús, J.A. Fat and lean masses in youths with Down syndrome: Gender differences. Res. Dev. Disabil. 2011, 32, 1685–1693. [Google Scholar] [CrossRef]
- Loveday, S.J.; Thompson, J.M.; Mitchell, E.A. Bioelectrical impedance for measuring percentage body fat in young persons with Down syndrome: Validation with dual-energy absorptiometry. Acta Paediatr. 2012, 101, e491–e495. [Google Scholar] [CrossRef]
- Osaili, T.M.; Attlee, A.; Naveed, H.; Maklai, H.; Mahmoud, M.; Hamadeh, N.; Asif, T.; Hasan, H.; Obaid, R.S. Physical Status and Parent-Child Feeding Behaviours in Children and Adolescents with Down Syndrome in The United Arab Emirates. Int. J. Environ. Res. Public Health 2019, 16, 2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, M.O.; Shea, C.O.; Gibson, L.; Leo, J.; Carty, C. The prevalence of obesity in children and young people with Down syndrome. J. Appl. Res. Intellect. Disabil. 2018, 31, 1225–1229. [Google Scholar] [CrossRef]
- Buonuomo, P.S.; Bartuli, A.; Mastrogiorgio, G.; Vittucci, A.; Di Camillo, C.; Bianchi, S.; Marafon, D.P.; Villani, A.; Valentini, D. Lipid profiles in a large cohort of Italian children with Down syndrome. Eur. J. Med Genet. 2016, 59, 392–395. [Google Scholar] [CrossRef] [PubMed]
- De Asua, D.R.; Parra, P.; Costa, R.; Moldenhauer, F.; Suarez, C. A Cross-Sectional Study of the Phenotypes of Obesity and Insulin Resistance in Adults with Down Syndrome. Diabetes Metab. J. 2014, 38, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Piedra, M.J.; Alberti, G.; Cerda, J.; Cárdenas, A.; Paul, M.A.; Lizama, M. Alta frecuencia de dislipidemias en niños y adolescentes con Síndrome de Down. Revista Chilena de Pediatría 2017, 88, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Pitchford, E.A.; Adkins, C.; Hasson, R.E.; Hornyak, J.E.; Ulrich, D.A. Association between Physical Activity and Adiposity in Adolescents with Down Syndrome. Med. Sci. Sports Exerc. 2018, 50, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, G.V.; Pereira, F.D.; Fernhall, B. Reduced exercise capacity in persons with Down syndrome: Cause, effect, and management. Ther. Clin. Risk Manag. 2010, 6, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Cockerill, C.C.; Frisch, C.D.; Rein, S.E.; Orvidas, L.J. Supraglottoplasty outcomes in children with Down syndrome. Int. J. Pediatr. Otorhinolaryngol. 2016, 87, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Rotmensch, S.; Goldstein, I.; Liberati, M.; Shalev, J.; Ben-Rafael, Z.; Copel, J.A. Fetal transcerebellar diameter in down syndrome. Obstet. Gynecol. 1997, 89, 534–537. [Google Scholar] [CrossRef]
- Guidi, S.; Ciani, E.; Bonasoni, P.; Santini, D.; Bartesaghi, R. Widespread Proliferation Impairment and Hypocellularity in the Cerebellum of Fetuses with Down Syndrome. Brain Pathol. 2010, 21, 361–373. [Google Scholar] [CrossRef]
- Rigoldi, C.; Galli, M.; Mainardi, L.; Crivellini, M.; Albertini, G. Postural control in children, teenagers and adults with Down syndrome. Res. Dev. Disabil. 2011, 32, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Paul, Y.; Ellapen, T.J.; Barnard, M.; Hammill, H.V.; Swanepoel, M. The health benefits of exercise therapy for patients with Down syndrome: A systematic review. Afr. J. Disabil. 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Bertapelli, F.; Pitetti, K.; Agiovlasitis, S.; Guerra-Junior, G. Overweight and obesity in children and adolescents with Down syndrome—prevalence, determinants, consequences, and interventions: A literature review. Res. Dev. Disabil. 2016, 57, 181–192. [Google Scholar] [CrossRef]
- Lubkowska, A.; Radecka, A.; Bryczkowska, I.; Rotter, I.; Laszczyńska, M.; Dudzińska, W. Serum Adiponectin and Leptin Concentrations in Relation to Body Fat Distribution, Hematological Indices and Lipid Profile in Humans. Int. J. Environ. Res. Public Health 2015, 12, 11528–11548. [Google Scholar] [CrossRef] [Green Version]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Löllmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.L.; Ravussin, E.; Pratley, R.E.; Lee, G.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Proto, C.; Romualdi, D.; Cento, R.M.; Romano, C.; Campagna, G.; Lanzone, A. Free and total leptin serum levels and soluble leptin receptors levels in two models of genetic obesity: The Prader-Willi and the Down syndromes. Metab. 2007, 56, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Magge, S.N.; O’Neill, K.L.; Shults, J.; Stallings, V.A.; Stettler, N. Leptin Levels among Prepubertal Children with Down Syndrome Compared with Their Siblings. J. Pediatr. 2008, 152, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsi, M.M.; Dogliotti, G.; Pedroni, F.; Galliera, E.; Malavazos, A.E.; Villa, R.; Chiappelli, M.; Licastro, F. Adipocytokines in Down’s syndrome, an atheroma-free model: Role of adiponectin. Arch. Gerontol. Geriatr. 2009, 48, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Yahia, S.; El-Farahaty, R.M.; El-Hawary, A.K.; El-Hussiny, M.A.; Abdel-Maseih, H.; El-Dahtory, F.; El-Gilany, A.-H. Leptin, insulin and thyroid hormones in a cohort of Egyptian obese Down syndrome children: A comparative study. BMC Endocr. Disord. 2012, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fructuoso, M.; Rachdi, L.; Philippe, E.; Denis, R.; Magnan, C.; Le Stunff, H.; Janel, N.; Dierssen, M. Increased levels of inflammatory plasma markers and obesity risk in a mouse model of Down syndrome. Free. Radic. Biol. Med. 2018, 114, 122–130. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Yamauchi, T.; Kubota, T.; Moroi, M.; Matsui, J.; Eto, K.; Yamashita, T.; Kamon, J.; Satoh, H.; et al. Disruption of Adiponectin Causes Insulin Resistance and Neointimal Formation. J. Biol. Chem. 2002, 277, 25863–25866. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Folco, E.J.; Minami, M.; Wara, A.; Feinberg, M.W.; Sukhova, G.K.; Colvin, R.A.; Kihara, S.; Funahashi, T.; Luster, A.D.; et al. Adiponectin Inhibits the Production of CXC Receptor 3 Chemokine Ligands in Macrophages and Reduces T-Lymphocyte Recruitment in Atherogenesis. Circ. Res. 2008, 102, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Nakamura, T.; Nishida, M.; Kumada, M.; Okamoto, Y.; Ohashi, K.; Nagaretani, H.; Kishida, K.; et al. Reciprocal Association of C-Reactive Protein With Adiponectin in Blood Stream and Adipose Tissue. Circulation 2003, 107, 671–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Enrich, B.; Tilg, H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem. Biophys. Res. Commun. 2004, 323, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Tenneti, N.; Dayal, D.; Sharda, S.; Panigrahi, I.; Didi, M.; Attri, S.V.; Sachdeva, N.; Bhalla, A.K. Concentrations of leptin, adiponectin and other metabolic parameters in non-obese children with Down syndrome. J. Pediatr. Endocrinol. Metab. 2017, 30, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nat. Cell Biol. 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, S.D.; Klier, K.; Braun, S.; Azen, C.; Geffner, M.E.; Buchanan, T.A. Obese Adolescents Show Impaired Meal Responses of the Appetite-Regulating Hormones Ghrelin and PYY. Obesity 2010, 18, 918–925. [Google Scholar] [CrossRef]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeöld, A.; Bianco, A.C. Cellular and Molecular Basis of Deiodinase-Regulated Thyroid Hormone Signaling1. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef] [Green Version]
- Chin, W.W.; Carr, F.E.; Burnside, J.; Darling, D.S. Thyroid Hormone Regulation of Thyrotropin Gene Expression. Recent Prog Horm. Res. 1993, 48, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Bakal, K.; Minokoshi, Y.; Hollenberg, A.N. Leptin Signaling Targets the Thyrotropin-Releasing Hormone Gene Promoterin Vivo. Endocrinology 2004, 145, 2221–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzoros, C.S.; Ozata, M.; Negrao, A.B.; Suchard, M.A.; Ziotopoulou, M.; Caglayan, S.; Elashoff, R.M.; Cogswell, R.J.; Negro, P.; Victoria, L.; et al. Synchronicity of Frequently Sampled Thyrotropin (TSH) and Leptin Concentrations in Healthy Adults and Leptin-Deficient Subjects: Evidence for Possible Partial TSH Regulation by Leptin in Humans. J. Clin. Endocrinol. Metab. 2001, 86, 3284–3291. [Google Scholar] [CrossRef]
- Oppenheimer, J.H.; Schwartz, H.L.; Lane, J.T.; Thompson, M.P. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J. Clin. Investig. 1991, 87, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Tian, Y.; Guo, M.; Liu, J.; Heng, D.; Zhu, B.; Yang, Y.; Zhang, C. Regulation of glucose transport by thyroid hormone in rat ovary. Cell Tissue Res. 2016, 366, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Nedvidkova, J.; Haluzik, M.; Bartak, V.; Dostalova, I.; Vlcek, P.; Racek, P.; Taus, M.; Behanova, M.; Svacina, S.; Alesci, S.; et al. Changes of Noradrenergic Activity and Lipolysis in the Subcutaneous Abdominal Adipose Tissue of Hypo- and Hyperthyroid Patients: AnIn VivoMicrodialysis Study. Ann. New York Acad. Sci. 2004, 1018, 541–549. [Google Scholar] [CrossRef]
- Hayek, A. Unimpaired Gluconeogenesis in Congenital Hypothyroidism. Horm. Metab. Res. 1979, 11, 256. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Fort, P.; Lifshitz, F.; Bellisario, R.; Davis, J.; Lanes, R.; Pugliese, M.; Richman, R.; Post, E.; David, R. Abnormalities of thyroid function in infants with Down syndrome. J. Pediatr. 1984, 104, 545–549. [Google Scholar] [CrossRef]
- Graber, E.; Chacko, E.; Regelmann, M.O.; Costin, G.; Rapaport, R. Down Syndrome and Thyroid Function. Endocrinol. Metab. Clin. North Am. 2012, 41, 735–745. [Google Scholar] [CrossRef]
- Pierce, M.J.; LaFranchi, S.H.; Pinter, J.D. Characterization of Thyroid Abnormalities in a Large Cohort of Children with Down Syndrome. Horm. Res. Paediatr. 2017, 87, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuoghi, O.A.; Topolski, F.; De Faria, L.P.; Occhiena, C.M.; Ferreira, N.D.S.P.; Ferlin, C.R.; De Mendonça, M.R. Prevalence of Dental Anomalies in Permanent Dentition of Brazilian Individuals with Down Syndrome. Open Dent. J. 2016, 10, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Pisacane, A.; Toscano, E.; Pirri, I.; Continisio, P.; Andria, G.; Zoli, B.; Strisciuglio, P.; Concolino, D.; Piccione, M.; Giudice, C.L.; et al. Down syndrome and breastfeeding. Acta Paediatr. 2003, 92, 1479–1481. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Bablani, D.; Chowdhry, A.; Thapar, R.; Gupta, P.; Mishra, S. Dentofacial and Cranial Changes in Down Syndrome. Osong Public Health Res. Perspect. 2014, 5, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.M.; Winer, R.A. Dental and Facial Characteristics in Down’s Syndrome (Mongolism). J. Dent. Res. 1965, 44, 197–208. [Google Scholar] [CrossRef]
- Anders, P.L.; Davis, E.L. Oral health of patients with intellectual disabilities: A systematic review. Spéc. Care Dent. 2010, 30, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Borodina, G.; Morozov, S. Children With Gastroesophageal Reflux Disease Consume More Calories and Fat Compared to Controls of Same Weight and Age. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 808–814. [Google Scholar] [CrossRef]
- Zarate, N.; Mearin, F.; Hidalgo, A.M.; Malagelada, J.-R. Prospective evaluation of esophageal motor dysfunction in Down’s syndrome. Am. J. Gastroenterol. 2001, 96, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Alagiakrishnan, K.; Bhanji, R.A.; Kurian, M. Evaluation and management of oropharyngeal dysphagia in different types of dementia: A systematic review. Arch. Gerontol. Geriatr. 2013, 56, 1–9. [Google Scholar] [CrossRef]
- Field, D.; Garland, M.; Williams, K. Correlates of specific childhood feeding problems. J. Paediatr. Child Health 2003, 39, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Nordstrøm, M.; Paus, B.; Andersen, L.F.; Kolset, S.O. Dietary aspects related to health and obesity in Williams syndrome, Down syndrome, and Prader–Willi syndrome. Food Nutr. Res. 2015, 59, 25487. [Google Scholar] [CrossRef] [Green Version]
- Huggard, D.; Kelly, L.; Ryan, E.; McGrane, F.; Lagan, N.; Roche, E.; Balfe, J.; Leahy, T.R.; Franklin, O.; Doherty, D.G.; et al. Increased systemic inflammation in children with Down syndrome. Cytokine 2020, 127, 154938. [Google Scholar] [CrossRef]
- Patel, S.R. Obstructive Sleep Apnea. Ann. Intern. Med. 2019, 171, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Skatrud, J.; Peppard, P.E. Risk Factors for Obstructive Sleep Apnea in Adults. JAMA 2004, 291, 2013–2016. [Google Scholar] [CrossRef]
- Phillips, B.G.; Hisel, T.M.; Kato, M.; Pesek, C.A.; Dyken, M.E.; Narkiewicz, K.; Somers, V.K. Recent weight gain in patients with newly diagnosed obstructive sleep apnea. J. Hypertens. 1999, 17, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Bixler, E.O.; Vgontzas, A.N.; Lin, H.-M.; Have, T.T.; Leiby, B.E.; Vela-Bueno, A.; Kales, A. Association of hypertension and sleep-disordered breathing. Arch. Intern. Med. 2000, 160, 2289–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.R.; Palmer, L.J.; Larkin, E.K.; Jenny, N.S.; White, D.P.; Redline, S. Relationship between Obstructive Sleep Apnea and Diurnal Leptin Rhythms. Sleep 2004, 27, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Lavie, L. Oxidative Stress—A Unifying Paradigm in Obstructive Sleep Apnea and Comorbidities. Prog. Cardiovasc. Dis. 2009, 51, 303–312. [Google Scholar] [CrossRef]
- Unnikrishnan, D.; Jun, J.; Polotsky, V. Inflammation in sleep apnea: An update. Rev. Endocr. Metab. Disord. 2015, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Maris, M.; Verhulst, S.; Wojciechowski, M.; Van De Heyning, P.; Boudewyns, A. Prevalence of Obstructive Sleep Apnea in Children with Down Syndrome. Sleep 2016, 39, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trois, M.S.; Capone, G.T.; Lutz, J.A.; Melendres, M.C.; Schwartz, A.R.; Collop, N.A.; Marcus, C.L. Obstructive Sleep Apnea in Adults with Down Syndrome. J. Clin. Sleep Med. 2009, 5, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, C.L.; Brooks, L.J.; Ward, S.D.; Draper, K.A.; Gozal, D.; Halbower, A.C.; Jones, J.; Lehmann, C.; Schechter, M.S.; Sheldon, S.H.; et al. Diagnosis and Management of Childhood Obstructive Sleep Apnea Syndrome. Pediatrics 2012, 130, e714–e755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, C.E.; Cubbin, C. Socioeconomic Status and Childhood Obesity: A Review of Literature from the Past Decade to Inform Intervention Research. Curr. Obes. Rep. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fornieles, G.; Camacho-Molina, A.; Rosety, M.A.; Díaz, A.J.; Rosety, I.; Rosety-Rodríguez, M.; Alvero-Cruz, J.R.; Rosety, M.; Ordonez, F.J. Maternal fat mass may predict overweight/obesity in non-instituzionalized women with intellectual disability. Nutrición Hospitalaria 2013, 28, 1918–1921. [Google Scholar] [PubMed]
- O’Neill, K.L.; Shults, J.; Stallings, V.A.; Stettler, N. Child-feeding practices in children with down syndrome and their siblings. J. Pediatr. 2005, 146, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, M.L.; McDougle, C.J. Pharmacotherapy of Down syndrome. Expert Opin. Pharmacother. 2018, 19, 1875–1889. [Google Scholar] [CrossRef]
- Dent, R.; Blackmore, A.; Peterson, J.; Habib, R.; Kay, G.P.; Gervais, A.; Taylor, V.; Wells, G. Changes in Body Weight and Psychotropic Drugs: A Systematic Synthesis of the Literature. PLoS ONE 2012, 7, e36889. [Google Scholar] [CrossRef] [Green Version]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.M.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef] [PubMed]
- Flament-Durand, J. The hypothalamus: Anatomy and functions. Acta Psychiatr. Belg. 1980, 80, 364–375. [Google Scholar]
- Clark, J.T.; Kalra, P.S.; Crowley, W.R.; Kalra, S.P. Neuropeptide y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 1984, 115, 427–429. [Google Scholar] [CrossRef]
- Panksepp, J.; Reilly, P.; Bishop, P.; Meeker, R.B.; Vilberg, T.R.; Kastin, A.J. Effects of α-MSH on motivation, vigilance and brain respiration. Pharmacol. Biochem. Behav. 1976, 5, 59–64. [Google Scholar] [CrossRef]
- Kristensen, P.; Judge, M.E.; Thim, L.; Ribel, U.; Christjansen, K.N.; Wulff, B.S.; Clausen, J.T.; Jensen, P.B.; Madsen, O.D.; Vrang, N.; et al. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nat. Cell Biol. 1998, 393, 72–76. [Google Scholar] [CrossRef]
- Leibowitz, S.F.; Hammer, N.J.; Chang, K. Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat. Physiol. Behav. 1981, 27, 1031–1040. [Google Scholar] [CrossRef]
- Mayer, J.; Thomas, D.W. Regulation of Food Intake and Obesity. Science 1967, 156, 328–337. [Google Scholar] [CrossRef]
- Elias, C.F.; Saper, C.B.; Maratos-Flier, E.; Tritos, N.A.; Lee, C.; Kelly, J.; Tatro, J.B.; Hoffman, G.E.; Ollmann, M.M.; Barsh, G.S.; et al. Chemically Defined Projections Linking the Mediobasal Hypothalamus and the Lateral Hy-pothalamic Area. J. Comp. Neurol. 1998, 402, 442–459. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; Bobinski, M. Hypothalamic abnormalities in Down syndrome. Prog. Clin. Boil. Res. 1991, 373, 153–167. [Google Scholar]
- Ebaik, J.-H. Dopamine Signaling in reward-related behaviors. Front. Neural Circuits 2013, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Palmiter, R.D. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007, 30, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.M.; Avena, N.M.; Gearhardt, A.N. Which Foods May Be Addictive? The Roles of Processing, Fat Content, and Glycemic Load. PLoS ONE 2015, 10, e0117959. [Google Scholar] [CrossRef]
- Yates, C.; Simpson, J.; Gordon, A.; Maloney, A.; Allison, Y.; Ritchie, I.; Urquhart, A. Catecholamines and cholinergic enzymes in pre-senile and senile Alzheimer-type dementia and down’s syndrome. Brain Res. 1983, 280, 119–126. [Google Scholar] [CrossRef]
- Reynolds, G.; Godridge, H. Alzheimer-like brain monoamine deficits in adults with down’s syndrome. Lancet 1985, 326, 1368–1369. [Google Scholar] [CrossRef]
- Martí, E.; Altafaj, X.; Dierssen, M.; De La Luna, S.; Fotaki, V.; Alvarez, M.; Pérez-Riba, M.; Ferrer, I.; Estivill, X. Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 2003, 964, 250–263. [Google Scholar] [CrossRef]
- O’Connor, E.C.; Kremer, Y.; Lefort, S.; Harada, M.; Pascoli, V.J.; Rohner, C.; Lüscher, C. Accumbal D1R Neurons Projecting to Lateral Hypothalamus Authorize Feeding. Neuron 2015, 88, 553–564. [Google Scholar] [CrossRef]
- London, J.; Rouch, C.; Bui, L.C.; Assayag, E.; Souchet, B.; Daubigney, F.; Medjaoui, H.; Luquet, S.; Magnan, C.; Delabar, J.M.; et al. Overexpression of the DYRK1A Gene (Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A) Induces Alterations of the Serotoninergic and Dopaminergic Processing in Murine Brain Tissues. Mol. Neurobiol. 2017, 55, 1–10. [Google Scholar] [CrossRef]
- Milunsky, A.; Neurath, P.W. Diabetes Mellitus in Down’s Syndrome. Arch. Environ. Health Int. J. 1968, 17, 372–376. [Google Scholar] [CrossRef]
- Jeremiah, D.E.; Leyshon, G.E.; Rose, T.; Francis, T.R.H.W.S.; Elliott, R.W. Down’s syndrome and diabetes. Psychol. Med. 1973, 3, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Bergholdt, R.; Eising, S.; Nerup, J.; Pociot, F. Increased prevalence of Down’s syndrome in individuals with type 1 diabetes in Denmark: A nationwide population-based study. Diabetol. 2006, 49, 1179–1182. [Google Scholar] [CrossRef] [Green Version]
- Burch, P.; Milunsky, A. Early-onset diabetes mellitus in the general and down’s syndrome populations. Lancet 1969, 293, 554–558. [Google Scholar] [CrossRef]
- Carnicer, J.; Farré, C.; Varea, V.; Vilar, P.; Moreno, J.; Artigas, J. Prevalence of coeliac disease in Down’s syndrome. Eur. J. Gastroenterol. Hepatol. 2001, 13, 263–267. [Google Scholar] [CrossRef]
- Yang, Q.; Rasmussen, S.A.; Friedman, J.M. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: A population-based study. Lancet 2002, 359, 1019–1025. [Google Scholar] [CrossRef]
- Selikowitz, M. Health problems and health checks in school-aged children with Down syndrome. J. Paediatr. Child Health 1992, 28, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.V.; Mahmoud, S.A.; Chen, H.; Lowery-Nordberg, M.; Berlin, K.; Bahna, S.L. Follow-up study of immune defects in patients with dysmorphic disorders. Ann. Allergy, Asthma Immunol. 2009, 102, 426–431. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; Von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRocca, L.M.; Lauriola, L.; Ranelletti, F.O.; Piantelli, M.; Maggiano, N.; Ricci, R.; Capelli, A. Morphological and immunohistochemical study of Down syndrome thymus. Am. J. Med Genet. 2005, 37, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.A.; Moreira-Filho, C.A.; Ramos, P.L.; Brentani, H.; Lima, L.D.A.; Arrais, M.; Bento-De-Souza, L.C.; Bento-De-Souza, L.; Duarte, M.I.; Coutinho, A.; et al. Decreased AIRE Expression and Global Thymic Hypofunction in Down Syndrome. J. Immunol. 2011, 187, 3422–3430. [Google Scholar] [CrossRef] [Green Version]
- Skogberg, G.; Lundberg, V.; Lindgren, S.; Gudmundsdottir, J.; Sandström, K.; Kämpe, O.; Annerén, G.; Gustafsson, J.; Sunnegårdh, J.; Van Der Post, S.; et al. Altered Expression of Autoimmune Regulator in Infant Down Syndrome Thymus, a Possible Contributor to an Autoimmune Phenotype. J. Immunol. 2014, 193, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Noble, J.A.; Valdes, A.M.; Varney, M.D.; Carlson, J.A.; Moonsamy, P.; Fear, A.L.; Lane, J.A.; Lavant, E.; Rappner, R.; Louey, A.; et al. HLA Class I and Genetic Susceptibility to Type 1 Diabetes: Results From the Type 1 Diabetes Genetics Consortium. Diabetes 2010, 59, 2972–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, R.J.; Mehers, K.L.; Williams, A.J.; Brown, J.; Bingley, P.J.; Holl, R.W.; Rohrer, T.R.; Schober, E.; Abdul-Rasoul, M.M.; Shield, J.P.; et al. Early-Onset, Coexisting Autoimmunity and Decreased HLA-Mediated Susceptibility Are the Characteristics of Diabetes in Down Syndrome. Diabetes Care 2012, 36, 1181–1185. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Satot, M.; Sasaki, A.; Ho, L.; Eckman, C.B.; Prada, C.-M.; Younkin, S.G.; Kobayashi, T.; Tada, N.; et al. Accumulation of β-Amyloid fibrils in pancreas of transgenic mice. Neurobiol. Aging 1996, 17, 215–222. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; Malester, B.; Lafrancois, J.; Zehr, C.; Daeschner, J.M.; Olschowka, J.A.; Fonseca, M.I.; O’Banion, M.K.; Tenner, A.J.; et al. Inflammatory Responses to Amyloidosis in a Transgenic Mouse Model of Alzheimer’s Disease. Am. J. Pathol. 2001, 158, 1345–1354. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurol. 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Insoft, R.M.; Pike-Nobile, L.; Epstein, L.B. A hypothesis to explain the immune defects in Down syndrome. Prog. Clin. Boil. Res. 1995, 393, 147–167. [Google Scholar]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [Green Version]
- Magge, S.N.; Zemel, B.S.; Pipan, M.E.; Gidding, S.S.; Kelly, A. Cardiometabolic Risk and Body Composition in Youth With Down Syndrome. Pediatrics 2019, 144, e20190137. [Google Scholar] [CrossRef]
- Broers, C.J.M.; Gemke, R.J.B.J.; Weijerman, M.E.; Van Der Sluijs, K.F.; Van Furth, A.M. Increased Pro-inflammatory Cytokine Production in Down syndrome Children Upon Stimulation with Live Influenza A Virus. J. Clin. Immunol. 2012, 32, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.; Debom, G.; Soares, F.; Machado, C.; Pureza, J.; Peres, W.; Garcias, G.D.L.; Duarte, M.F.; Schetinger, M.R.C.; Stefanello, F.; et al. Alterations of ectonucleotidases and acetylcholinesterase activities in lymphocytes of Down syndrome subjects: Relation with inflammatory parameters. Clin. Chim. Acta 2014, 433, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cetiner, S.; Demirhan, O.; Inal, T.C.; Tastemir, D.; Sertdemir, Y. Analysis of peripheral blood T-cell subsets, natural killer cells and serum levels of cytokines in children with Down syndrome. Int. J. Immunogenetics 2010, 37, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Fonctional loss of pancreatic islets in type 2 diabetes: How can we halt it? Metabolism 2020, 110, 154304. [Google Scholar] [CrossRef]
- Sorci, G.; Agneletti, A.L.; Donato, R. Effects of S100A1 and S100B on microtubule stability. An in vitro study using triton-cytoskeletons from astrocyte and myoblast cell lines. Neuroscience 2000, 99, 773–783. [Google Scholar] [CrossRef]
- Gentil, B.J.; Delphin, C.; Mbele, G.O.; Deloulme, J.C.; Ferro, M.; Garin, J.; Baudier, J. The Giant Protein AHNAK Is a Specific Target for the Calcium- and Zinc-binding S100B Protein. J. Biol. Chem. 2001, 276, 23253–23261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcuri, C.; Bianchi, R.; Brozzi, F.; Donato, R. S100B Increases Proliferation in PC12 Neuronal Cells and Reduces Their Responsiveness to Nerve Growth Factor via Akt Activation. J. Biol. Chem. 2005, 280, 4402–4414. [Google Scholar] [CrossRef] [Green Version]
- Tubaro, C.; Arcuri, C.; Giambanco, I.; Donato, R. S100B protein in myoblasts modulates myogenic differentiation via NF-κB-dependent inhibition of MyoD expression. J. Cell. Physiol. 2010, 223, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, F.; Tsoporis, J.N.; Izhar, S.; Desjardins, J.-F.; Parker, T.G. Deficiency of S100B confers resistance to experimental diabetes in mice. Exp. Cell Res. 2018, 365, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Peiris, H.; Raghupathi, R.; Jessup, C.F.; Zanin, M.P.; Mohanasundaram, D.; MacKenzie, K.D.; Chataway, T.; Clarke, J.N.; Brealey, J.; Coates, P.T.; et al. Increased Expression of the Glucose-Responsive Gene, RCAN1, Causes Hypoinsulinemia, β-Cell Dysfunction, and Diabetes. Endocrinology 2012, 153, 5212–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helguera, P.; Seiglie, J.; Rodriguez, J.; Hanna, M.; Helguera, G.; Busciglio, J. Adaptive Downregulation of Mitochondrial Function in Down Syndrome. Cell Metab. 2013, 17, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.-Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; González, B.J.; Bender, A.; Takane, K.K.; et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 2019, 29, 638–652.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Cell Replication Is the Primary Mechanism Subserving the Postnatal Expansion of -Cell Mass in Humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.J. Linking the Genetics of Type 2 Diabetes With Low Birth Weight: A Role for Prenatal Islet Maldevelopment? Diabetes 2009, 58, 1255–1256. [Google Scholar] [CrossRef] [Green Version]
- Song, W.-J.; Song, E.-A.C.; Jung, M.-S.; Choi, S.-H.; Baik, H.-H.; Jin, B.K.; Kim, J.H.; Chung, S.-H. Phosphorylation and Inactivation of Glycogen Synthase Kinase 3β (GSK3β) by Dual-specificity Tyrosine Phosphorylation-regulated Kinase 1A (Dyrk1A). J. Biol. Chem. 2015, 290, 2321–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, J.; Kulkarni, R.N. Novel factors modulating human β-cell proliferation. Diabetes, Obes. Metab. 2016, 18, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Sacks, W.; Rizza, R.A.; Butler, P.C. Down Syndrome-Associated Diabetes Is Not Due To a Congenital Deficiency in β Cells. J. Endocr. Soc. 2017, 1, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Peiris, H.; Duffield, M.D.; Fadista, J.; Jessup, C.F.; Kashmir, V.; Genders, A.J.; McGee, S.L.; Martin, A.M.; Saiedi, M.; Morton, N.; et al. A Syntenic Cross Species Aneuploidy Genetic Screen Links RCAN1 Expression to β-Cell Mitochondrial Dysfunction in Type 2 Diabetes. PLoS Genet. 2016, 12, e1006033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ptomey, L.T.; Walpitage, D.L.; Mohseni, M.; Gillette, M.L.D.; Davis, A.M.; Forseth, B.; Dean, E.E.; Waitman, L.R. Weight status and associated comorbidities in children and adults with Down syndrome, autism spectrum disorder and intellectual and developmental disabilities. J. Intellect. Disabil. Res. 2020, 64, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreau, M.; Benhaddou, S.; Dard, R.; Tolu, S.; Hamzé, R.; Vialard, F.; Movassat, J.; Janel, N. Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines 2021, 9, 221. https://doi.org/10.3390/biomedicines9020221
Moreau M, Benhaddou S, Dard R, Tolu S, Hamzé R, Vialard F, Movassat J, Janel N. Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines. 2021; 9(2):221. https://doi.org/10.3390/biomedicines9020221
Chicago/Turabian StyleMoreau, Manon, Soukaina Benhaddou, Rodolphe Dard, Stefania Tolu, Rim Hamzé, François Vialard, Jamileh Movassat, and Nathalie Janel. 2021. "Metabolic Diseases and Down Syndrome: How Are They Linked Together?" Biomedicines 9, no. 2: 221. https://doi.org/10.3390/biomedicines9020221
APA StyleMoreau, M., Benhaddou, S., Dard, R., Tolu, S., Hamzé, R., Vialard, F., Movassat, J., & Janel, N. (2021). Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines, 9(2), 221. https://doi.org/10.3390/biomedicines9020221