
Profiling Inflammatory Extracellular Vesicles in Plasma and Cerebrospinal Fluid: An Optimized Diagnostic Model for Parkinson’s Disease

 ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
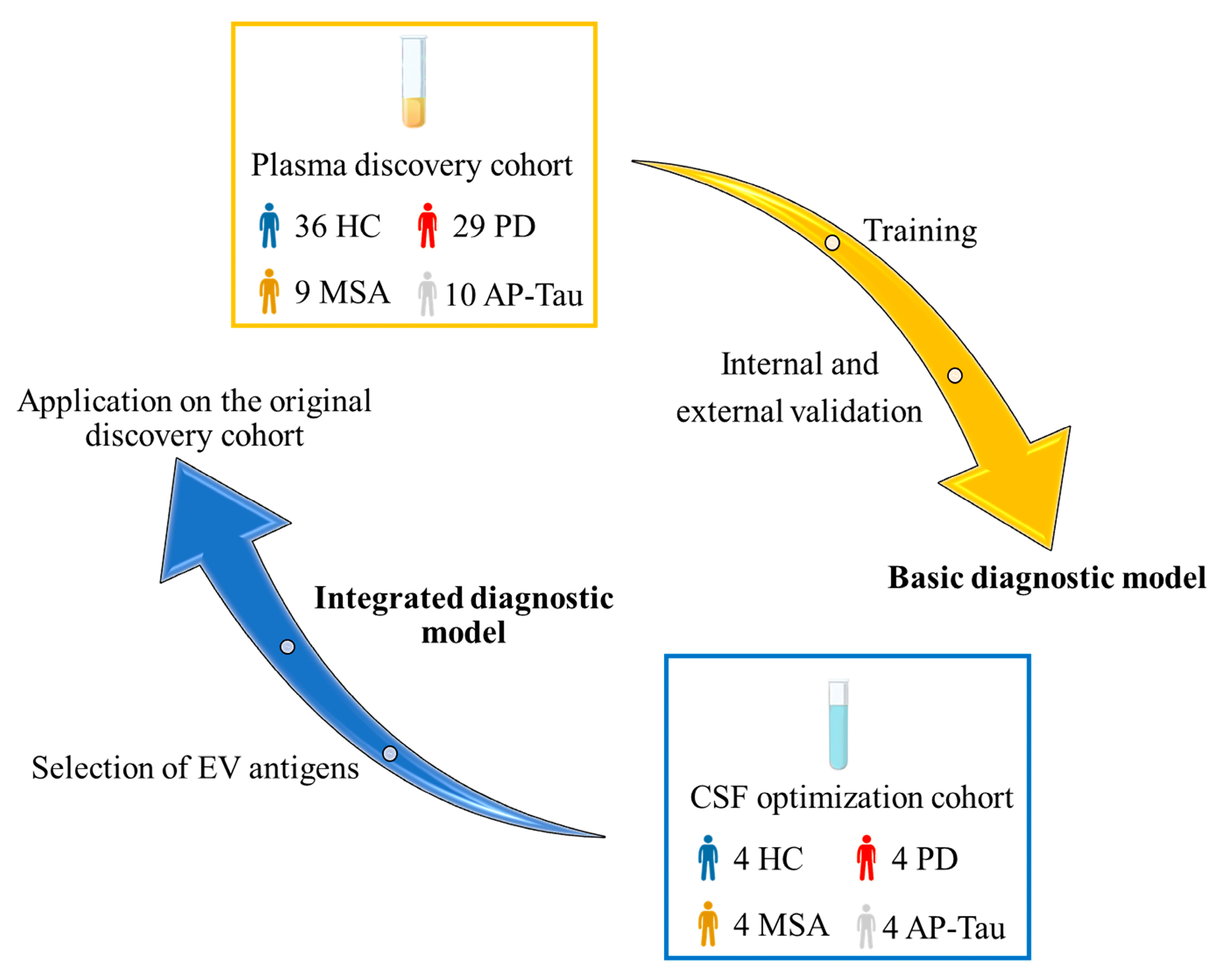
2.1. Subjects
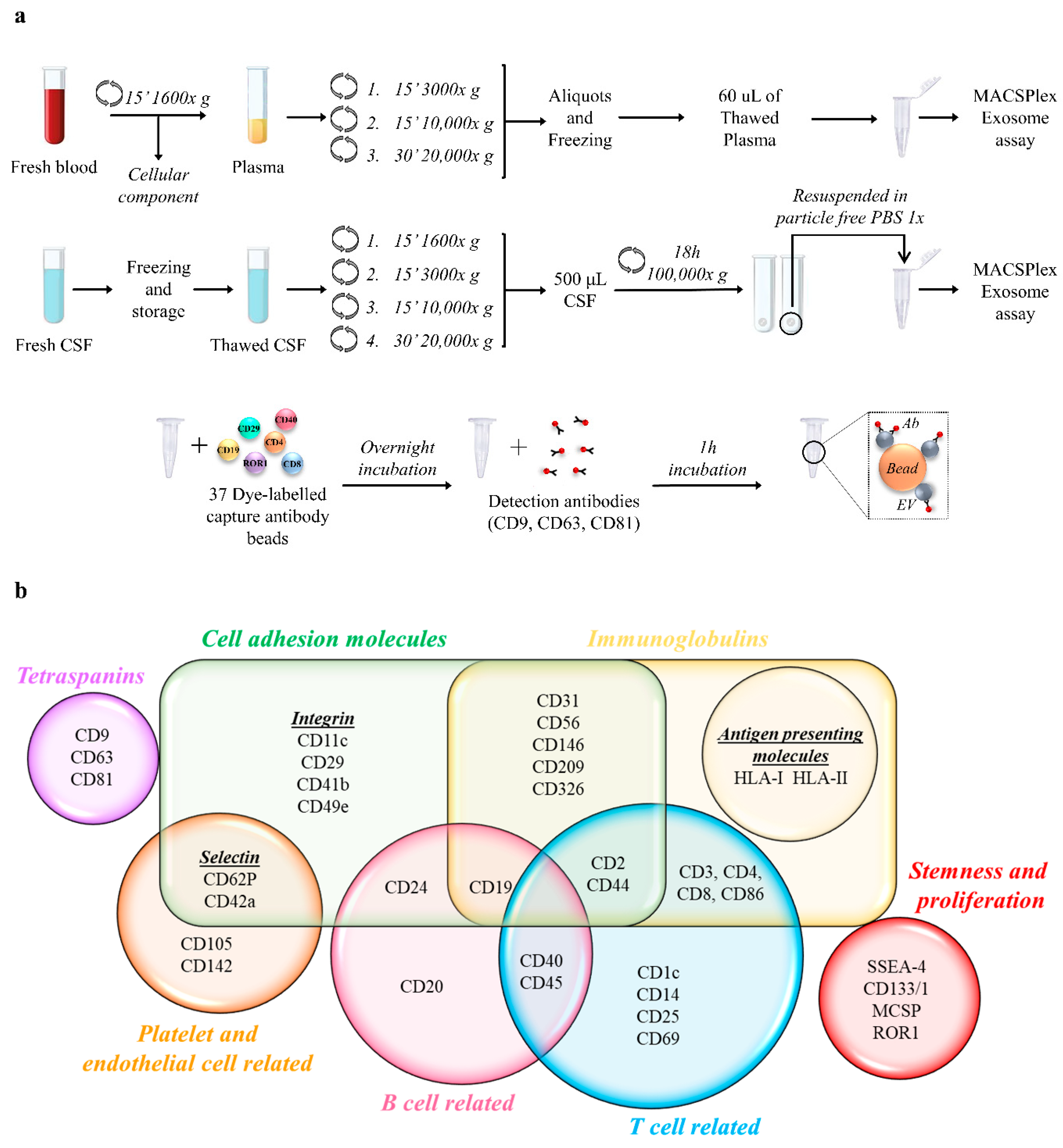
2.2. Plasma and CSF Preparation
2.3. Nanoparticle Tracking Analysis (NTA)
2.4. MACSPlex Exosome Assay and Flow Cytometry Analysis
2.5. Statistical Analysis
2.6. Diagnostic Modelling
3. Results
3.1. Demographic and Clinical Characteristics
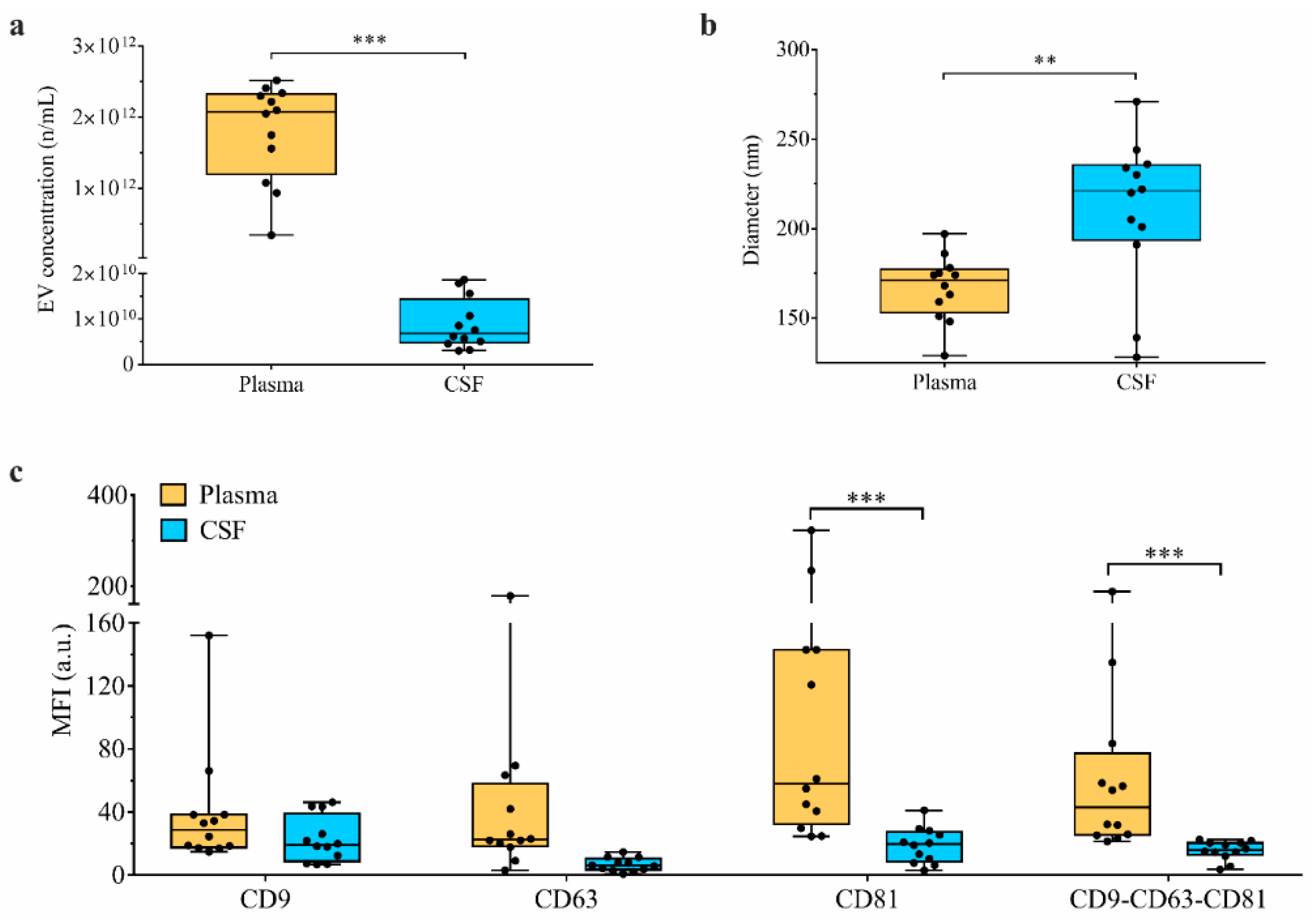
3.2. PD Group Shows an Increased Number of CSF-Derived EVs
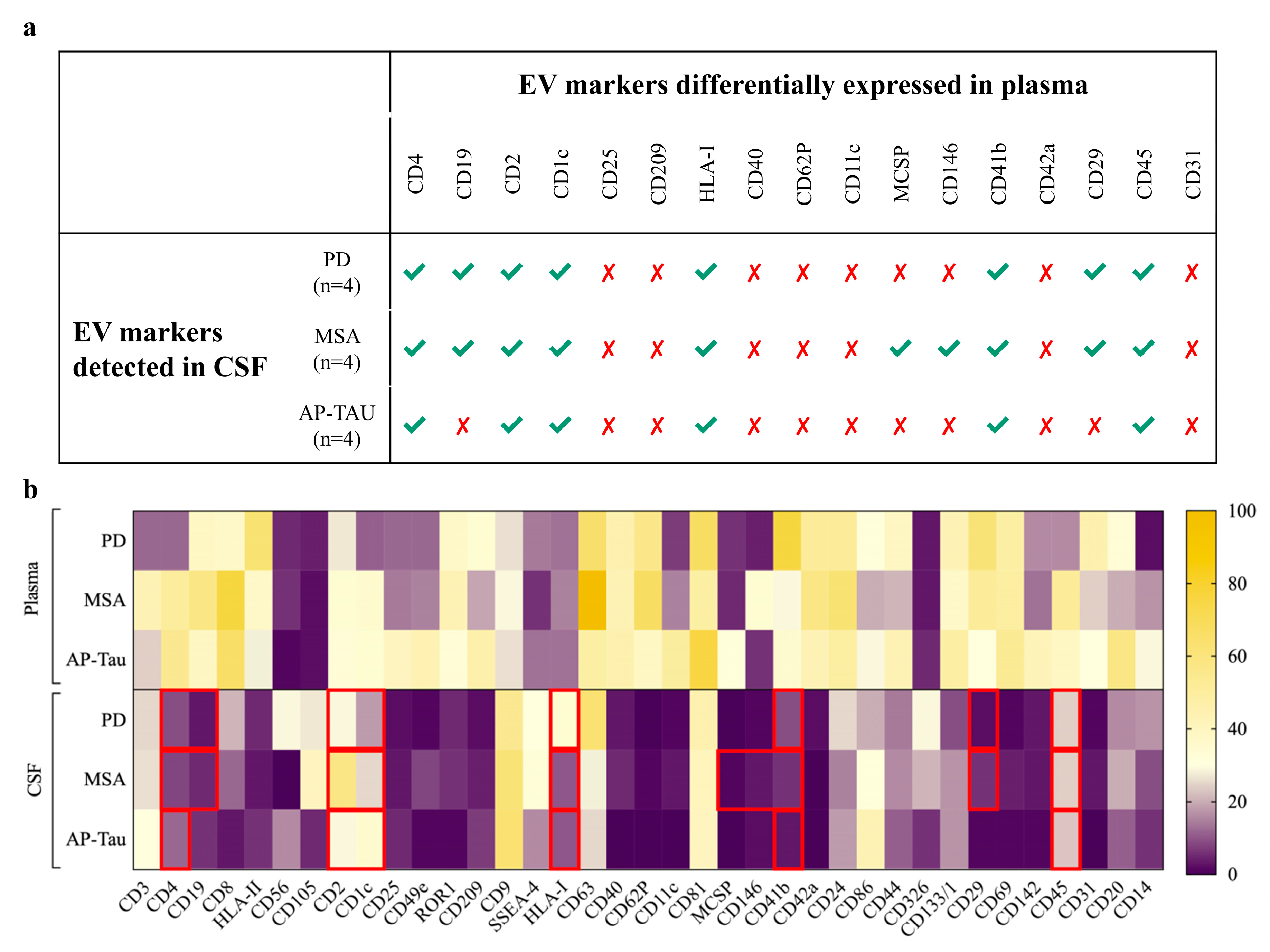
3.3. CSF-Derived EV Immunophenotyping Stratifies Patients According to the Clinical Diagnosis
3.4. Paired Analysis of CSF vs. Plasma in Patients Identifies Different Amount of EVs and Different Surface Marker Expressions
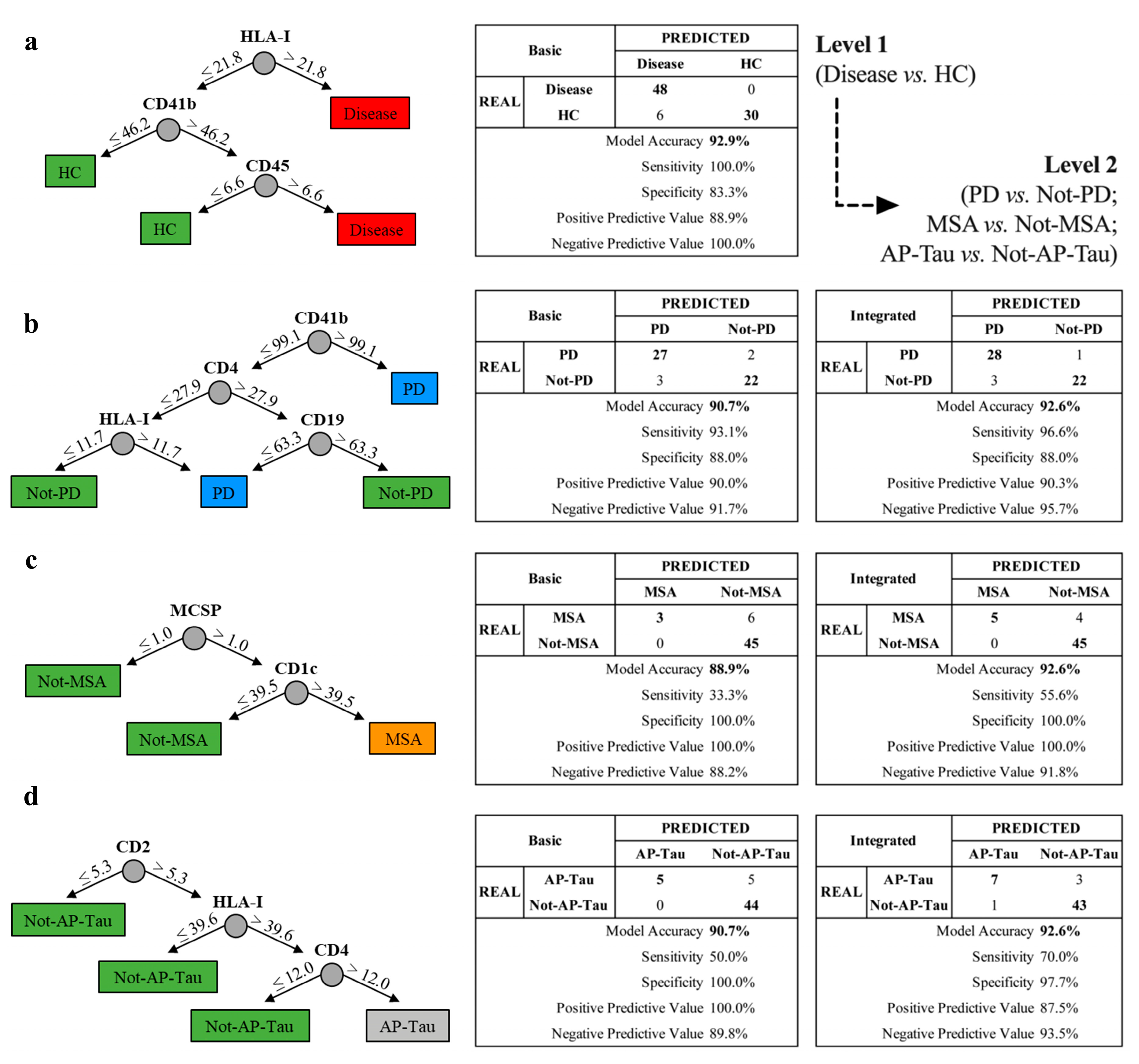
3.5. The Integrated Random Forest Model Demonstrates Higher Diagnostic Accuracy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Andjus, P.; Kosanovic, M.; Milicevic, K.; Gautam, M.; Vainio, S.J.; Jagevcic, D.; Kozlova, E.N.; Pivoriunas, A.; Chachques, J.; Sakaj, M.; et al. Extracellular Vesicles as Innovative Tool for Diagnosis, Regeneration and Protection against Neurological Damage. Int. J. Mol. Sci. 2020, 21, 6859. [Google Scholar] [CrossRef]
- Pinnell, J.R.; Cui, M.; Tieu, K. Exosomes in Parkinson Disease. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of Toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Koliha, N.; Heider, U.; Ozimkowski, T.; Wiemann, M.; Bosio, A.; Wild, S. Melanoma Affects the Composition of Blood Cell-Derived Extracellular Vesicles. Front. Immunol. 2016, 7, 282. [Google Scholar] [CrossRef] [Green Version]
- Koliha, N.; Wiencek, Y.; Heider, U.; Jungst, C.; Kladt, N.; Krauthauser, S.; Johnston, I.C.D.; Bosio, A.; Schauss, A.; Wild, S. A novel multiplex bead-based platform highlights the diversity of extracellular vesicles. J. Extracell. Vesicles 2016, 5, 29975. [Google Scholar] [CrossRef]
- Vacchi, E.; Burrello, J.; Di Silvestre, D.; Burrello, A.; Bolis, S.; Mauri, P.; Vassalli, G.; Cereda, C.W.; Farina, C.; Barile, L. Immune profiling of plasma-derived extracellular vesicles identifies Parkinson disease. Neurol Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Li, Q.; Lai, H.; Zhang, H.; Hu, Z.; Li, Y.; Huang, S. EV-origin: Enumerating the tissue-cellular origin of circulating extracellular vesicles using exLR profile. Comput. Struct. Biotechnol. J. 2020, 18, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Street, J.M.; Barran, P.E.; Mackay, C.L.; Weidt, S.; Balmforth, C.; Walsh, T.S.; Chalmers, R.T.A.; Webb, D.J.; Dear, J.W. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J. Transl. Med. 2012, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Gilman, S.; Wenning, G.K.; Low, P.A.; Brooks, D.J.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.W.; Colosimo, C.; Durr, A.; Fowler, C.J. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Muller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Kipfer, S.; Stephan, M.A.; Schupbach, W.M.; Ballinari, P.; Kaelin-Lang, A. Resting tremor in Parkinson disease: A negative predictor of levodopa-induced dyskinesia. Arch. Neurol. 2011, 68, 1037–1039. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 22. [Google Scholar] [CrossRef]
- Burrello, J.; Bolis, S.; Balbi, C.; Burrello, A.; Provasi, E.; Caporali, E.; Gauthier, L.G.; Peirone, A.; D’Ascenzo, F.; Monticone, S.; et al. An extracellular vesicle epitope profile is associated with acute myocardial infarction. J. Cell Mol. Med. 2020, 24, 9945–9957. [Google Scholar] [CrossRef]
- Castellani, C.; Burrello, J.; Fedrigo, M.; Burrello, A.; Bolis, S.; di Silvestre, D.; Tona, F.; Bottio, T.; Biemmi, V.; Toscano, G.; et al. Circulating extracellular vesicles as non-invasive biomarker of rejection in heart transplant. J. Heart Lung Transplant. 2020, 39, 1136–1148. [Google Scholar] [CrossRef]
- Agliardi, C.; Meloni, M.; Guerini, F.R.; Zanzottera, M.; Bolognesi, E.; Baglio, F.; Clerici, M. Oligomeric alpha-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson’s disease. Neurobiol. Dis. 2021, 148, 105185. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosta, F.; Dalla Libera, D.; Spinelli, E.G.; Finardi, A.; Canu, E.; Bergami, A.; Bocchio Chiavetto, L.; Baronio, M.; Comi, G.; Martino, G.; et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann. Neurol. 2014, 76, 813–825. [Google Scholar] [CrossRef]
- Verderio, C.; Muzio, L.; Turola, E.; Bergami, A.; Novellino, L.; Ruffini, F.; Riganti, L.; Corradini, I.; Francolini, M.; Garzetti, L.; et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012, 72, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Tietje, A.; Maron, K.N.; Wei, Y.; Feliciano, D.M. Cerebrospinal fluid extracellular vesicles undergo age dependent declines and contain known and novel non-coding RNAs. PLoS ONE 2014, 9, e113116. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Romero-Ramos, M. Immune system responses in Parkinson’s disease: Early and dynamic. Eur. J. Neurosci. 2019, 49, 364–383. [Google Scholar] [CrossRef]
- Reinert, K.R.; Umphlet, C.D.; Quattlebaum, A.; Boger, H.A. Short-term effects of an endotoxin on substantia nigra dopamine neurons. Brain Res. 2014, 1557, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Boza-Serrano, A.; Dunning, C.J.R.; Clausen, B.H.; Lambertsen, K.L.; Deierborg, T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflamm. 2018, 15, 168. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Slomka, A.; Urban, S.K.; Lukacs-Kornek, V.; Zekanowska, E.; Kornek, M. Large Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front. Immunol. 2018, 9, 2723. [Google Scholar] [CrossRef]
- Sankowski, R.; Mader, S.; Valdes-Ferrer, S.I. Systemic inflammation and the brain: Novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front. Cell Neurosci. 2015, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, J.L. Macrophages: Their Untold Story in T Cell Activation and Function. Int. Rev. Cell Mol. Biol. 2019, 342, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Stalter, R.D. Chapter 1—MHC Class I. In Measuring Immunity—Basic Biology and Clinical Assessment; Thomson, M., Ed.; Elsevier: New York, NY, USA, 2005; pp. 3–11. [Google Scholar]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bove, J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 2020, 143, 3717–3733. [Google Scholar] [CrossRef]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Rossi, E.; Bernabeu, C.; Smadja, D.M. Endoglin as an Adhesion Molecule in Mature and Progenitor Endothelial Cells: A Function Beyond TGF-beta. Front. Med. 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Lastres, P.; Bellon, T.; Cabanas, C.; Sanchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabeu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.F.; Beach, T.G.; Tooyama, I. Microglial Phenotyping in Neurodegenerative Disease Brains: Identification of Reactive Microglia with an Antibody to Variant of CD105/Endoglin. Cells 2019, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, I.; Peter, S.; Beyer, M.; Gimsa, U.; Nitsch, R. Presence of B7-2 (CD86) and lack of B7-1 (CD(80) on myelin phagocytosing MHC-II-positive rat microglia is associated with nondestructive immunity in vivo. FASEB J. 2001, 15, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Issazadeh, S.; Navikas, V.; Schaub, M.; Sayegh, M.; Khoury, S. Kinetics of expression of costimulatory molecules and their ligands in murine relapsing experimental autoimmune encephalomyelitis in vivo. J. Immunol. 1998, 161, 1104–1112. [Google Scholar]
- Zeinstra, E.; Wilczak, N.; De Keyser, J. Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7-1 and B7-2. J. Neuroimmunol. 2003, 135, 166–171. [Google Scholar] [CrossRef]
- Cesselli, D.; Parisse, P.; Aleksova, A.; Veneziano, C.; Cervellin, C.; Zanello, A.; Beltrami, A.P. Extracellular Vesicles: How Drug and Pathology Interfere With Their Biogenesis and Function. Front. Physiol. 2018, 9, 1394. [Google Scholar] [CrossRef]
- Szatanek, R.; Baran, J.; Siedlar, M.; Baj-Krzyworzeka, M. Isolation of extracellular vesicles: Determining the correct approach (Review). Int. J. Mol. Med. 2015, 36, 11–17. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | HC (n = 4) | PD (n = 4) | AP | p-Value | |
|---|---|---|---|---|---|
| MSA (n = 4) | AP-Tau (n = 4) | ||||
| Age (years) | 59 ± 20.3 | 67 ± 12.9 | 60 ± 4.6 | 66 ± 12.5 | 0.794 |
| Sex (ref. male) | 1 (25.0) | 2 (50.0) | 1 (25.0) | 2 (50.0) | 0.785 |
| Disease duration (years) | - | 4.0 [0.8–6.5] | 3.0 [1.0–5.8] | 3.5 [3.0–8.5] | 0.859 |
| H&Y | - | 1.5 [1.0–2.8] | 3.5 [2.3–4.8] | 4.0 [2.0–4.0] | 0.129 |
| MDS-UPDRS | - | 34.5 [17.0–51.3] | 34.8 [26.5–34.8] | 67.0 [29.0–67.0] | 0.511 |
| Beck Depression | - | 7.0 [2.3–14.0] | 5.5 [3.5–9.0] | 16.0 [11.0–16.0] | 0.094 |
| MMSE | - | 30.0 [30.0–30.0] | 28.0 [24.5–30.0] | 24.0 [22.0–24.0] | 0.051 |
| MOCA | - | 28.0 [26.3–29.8] | 23.0 [16.3–29.0] | 19.0 [16.0–19.0] | 0.114 |
| Olfactory test | - | 6.0 [3.0–6.0] | 9.0 [4.5–10.5] | 7.0 [5.0–7.0] | 0.722 |
| RBD | - | 7.0 [3.0–8.8] | 4.0 [2.5–4.8] | 2.0 [1.0–2.0] | 0.139 |
| LEDD | - | 238 [100.0–750.0] | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacchi, E.; Burrello, J.; Burrello, A.; Bolis, S.; Monticone, S.; Barile, L.; Kaelin-Lang, A.; Melli, G. Profiling Inflammatory Extracellular Vesicles in Plasma and Cerebrospinal Fluid: An Optimized Diagnostic Model for Parkinson’s Disease. Biomedicines 2021, 9, 230. https://doi.org/10.3390/biomedicines9030230
Vacchi E, Burrello J, Burrello A, Bolis S, Monticone S, Barile L, Kaelin-Lang A, Melli G. Profiling Inflammatory Extracellular Vesicles in Plasma and Cerebrospinal Fluid: An Optimized Diagnostic Model for Parkinson’s Disease. Biomedicines. 2021; 9(3):230. https://doi.org/10.3390/biomedicines9030230
Chicago/Turabian StyleVacchi, Elena, Jacopo Burrello, Alessio Burrello, Sara Bolis, Silvia Monticone, Lucio Barile, Alain Kaelin-Lang, and Giorgia Melli. 2021. "Profiling Inflammatory Extracellular Vesicles in Plasma and Cerebrospinal Fluid: An Optimized Diagnostic Model for Parkinson’s Disease" Biomedicines 9, no. 3: 230. https://doi.org/10.3390/biomedicines9030230
APA StyleVacchi, E., Burrello, J., Burrello, A., Bolis, S., Monticone, S., Barile, L., Kaelin-Lang, A., & Melli, G. (2021). Profiling Inflammatory Extracellular Vesicles in Plasma and Cerebrospinal Fluid: An Optimized Diagnostic Model for Parkinson’s Disease. Biomedicines, 9(3), 230. https://doi.org/10.3390/biomedicines9030230