
Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology

 ,
,  ,
,  , ,
, ,
Abstract
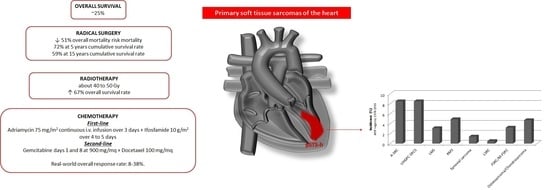
:
1. Introduction
2. Classification, Pathological Features, and Molecular Features
2.1. Angiosarcoma
2.2. Undifferentiated High-Grade Pleomorphic Sarcoma
2.3. Rhabdomyosarcoma
2.4. Leiomyosarcoma
2.5. Synovial Sarcoma
2.6. Liposarcoma
2.7. Fibrosarcoma and Myxoid Fibrosarcoma
2.8. Osteosarcoma and Chondrosarcoma
3. The Role of Echocardiography in pSTS-h
4. cMRI in pSTS-h
5. The Therapeutic Options
5.1. Surgical Treatment
5.2. Radiotherapy
5.3. Chemotherapy
6. New Therapeutic Strategies and Future Perspectives
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahouma, M.; Arisha, M.J.; Elmously, A.; Ahmed, M.M.E.-S.; Spadaccio, C.; Mehta, K.; Baudo, M.; Kamel, M.; Mansor, E.; Ruan, Y.; et al. Cardiac tumors prevalence and mortality: A systematic review and meta-analysis. Int. J. Surg. 2020, 76, 178–189. [Google Scholar] [CrossRef]
- Reynen, K. Frequency of primary tumors of the heart. Am. J. Cardiol. 1996, 77, 107. [Google Scholar] [CrossRef]
- McAllister, H.A.; Fenoglio, J.J. Tumors of the cardiovascular system. In Atlas of Tumor Pathology; Second Series, Fascicle 15; Hartmann, W.H., Cowan, W.R., Eds.; Armed Forces Institute of Pathology: Washington, DC, USA, 1978; pp. 1–3. [Google Scholar]
- Lestuzzi, C. Primary tumors of the heart. Curr. Opin. Cardiol. 2016, 31, 593–598. [Google Scholar] [CrossRef]
- Antwi-Amoabeng, D.; Meghji, Z.; Thakkar, S.; Ulanja, M.B.; Taha, M.; Adalja, D.; Al-Khafaji, J.; Gullapalli, N.; Beutler, B.D.; Boampong-Konam, K.; et al. Survival Differences in Men and Women With Primary Malignant Cardiac Tumor: An Analysis Using the Surveillance, Epidemiology and End Results (SEER) Database From 1973 to 2015. J. Am. Hear. Assoc. 2020, 9, e014846. [Google Scholar] [CrossRef]
- Mayer, F.; Aebert, H.; Rudert, M.; Königsrainer, A.; Horger, M.; Kanz, L.; Bamberg, M.; Ziemer, G.; Hartmann, J.T. Primary Malignant Sarcomas of the Heart and Great Vessels in Adult Patients—A Single-Center Experience. Oncologist 2007, 12, 1134–1142. [Google Scholar] [CrossRef]
- Orlandi, A.; Ferlosio, A.; Roselli, M.; Chiariello, L.; Spagnoli, L.G. Cardiac Sarcomas: An Update. J. Thorac. Oncol. 2010, 5, 1483–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isambert, N.; Ray-Coquard, I.; Italiano, A.; Rios, M.; Kerbrat, P.; Gauthier, M.; Blouet, A.; Chaigneau, L.; Duffaud, F.; Piperno-Neumann, S.; et al. Primary cardiac sarcomas: A retrospective study of the French Sarcoma Group. Eur. J. Cancer 2014, 50, 128–136. [Google Scholar] [CrossRef]
- Burke, A.P.; Virmani, R. Sarcomas of the great vessels. A clinicopathologic study. Cancer 1993, 71, 1761–1773. [Google Scholar] [CrossRef]
- Blackmon, S.H.; Patel, A.; Reardon, M.J. Management of primary cardiac sarcomas. Expert Rev. Cardiovasc. Ther. 2008, 6, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Reardon, M.J.; Walkes, J.-C.; Benjamin, R. Therapy Insight: Malignant primary cardiac tumors. Nat. Clin. Pract Neurol. 2006, 3, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Bendel, E.C.; Maleszewski, J.J.; Araoz, P.A. Imaging Sarcomas of the Great Vessels and Heart. Semin. Ultrasound CT MRI 2011, 32, 377–404. [Google Scholar] [CrossRef]
- Restrepo, C.S.; Vargas, D.; Ocazionez, D.; Martínez-Jiménez, S.; Cuellar, S.L.B.; Gutierrez, F.R. Primary Pericardial Tumors. Radiographics 2013, 33, 1613–1630. [Google Scholar] [CrossRef]
- Ostrowski, S.; Marcinkiewicz, A.; Kośmider, A.; Jaszewski, R. State of the art paper Sarcomas of the heart as a difficult interdisciplinary problem. Arch. Med. Sci. 2014, 1, 135–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, J.; Nakayama, J.; Yoshimura, Y.; Ikeda, U. Clinical classification of cardiovascular tumors and tumor-like lesions, and its incidences. Gen. Thorac. Cardiovasc. Surg. 2013, 61, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, A.; Tavora, F.R.; Maleszewski, J.J.; Frazier, A.A. Tumors of the heart and great vessels. In Atlas of Tumor Pathology; 4th Series; Armed Forces Institute of Pathology: Washington, DC, USA, 2014; pp. 291–336. Volume 22. [Google Scholar]
- Amano, J.; Nakayama, J. Epidemiology and frequency of cardiac tumors. In Textbook of Cardiac Tumors; Amano, J., Nakayama, J., Ikeda, U., Eds.; Nanzando: Tokyo, Japan, 2011; pp. 8–18. [Google Scholar]
- Grebenc, M.L.; De Christenson, M.L.R.; Burke, A.P.; Green, C.E.; Galvin, J.R. Primary Cardiac and Pericardial Neoplasms: Radiologic-Pathologic Correlation. Radiographics 2000, 20, 1073–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarjeant, J.M.; Butany, J.; Cusimano, R.J. Cancer of the heart: Epidemiology and management of primary tumors and metastases. Am. J. Cardiovasc. Drugs 2003, 3, 407–421. [Google Scholar] [CrossRef]
- Leduc, C.; Jenkins, S.M.; Sukov, W.R.; Rustin, J.G.; Maleszewski, J.J. Cardiac angiosarcoma: Histopathologic, immunohistochemical, and cytogenetic analysis of 10 cases. Hum. Pathol. 2017, 60, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Kassop, D.; Donovan, M.S.; Cheezum, M.K.; Nguyen, B.T.; Gambill, N.B.; Blankstein, R.; Villines, T.C. Cardiac Masses on Cardiac CT: A Review. Curr. Cardiovasc. Imaging Rep. 2014, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Neragi-Miandoab, S.; Kim, J.; Vlahakes, G. Malignant Tumours of the Heart: A Review of Tumour Type, Diagnosis and Therapy. Clin. Oncol. 2007, 19, 748–756. [Google Scholar] [CrossRef]
- Loukas, M.; Patel, S.D.; Peterson, A.; Bartczak, A.; Lee, S.; Chojnowski, S.; Gajewski, P. Primary cardiac angiosarcoma—A review. Med Sci. Monit. 2014, 20, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Park, H.K.; Jung, H.Y.; Lee, S.-Y.; Min, K.-W.; Kim, W.Y.; Han, H.S.; Kim, W.S.; Hwang, T.S.; Lim, S.D. ERG Immunohistochemistry as an Endothelial Marker for Assessing Lymphovascular Invasion. Korean J. Pathol. 2013, 47, 355–364. [Google Scholar] [CrossRef]
- Shanmugam, G. Primary cardiac sarcoma. Eur. J. Cardio-Thoracic Surg. 2006, 29, 925–932. [Google Scholar] [CrossRef]
- Salvador, C.; Saigí, M.; Díaz-Beveridge, R.; Penín, R.M.; Pané-Foix, M.; Mayordomo, E.; Melián, M.; Schuler, M.; Del Muro, X.G.; de Mora, J.F. Identification Of Actionable Genetic Targets In Primary Cardiac Sarcomas. OncoTargets Ther. 2019, 12, 9265–9275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raaf, H.N.; Raaf, J.H. Sarcomas related to the heart and vasculature. Semin. Surg. Oncol. 1994, 10, 374–382. [Google Scholar] [CrossRef]
- Burke, A. Primary malignant cardiac tumors. Semin. Diagn. Pathol. 2008, 25, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-G.; Cui, L.; Jiang, T.; Li, Y.-J.; Wei, Z.-M. Primary cardiac leiomyosarcoma: An analysis of clinical characteristics and outcome patterns. Asian Cardiovasc. Thorac. Ann. 2015, 23, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Blachman-Braun, R.; Aboitiz-Rivera, C.M.; Fraustro, A.A.; Ransom-Rodríguez, A.; Baltazares-Lipp, M.E.; Catrip-Torres, J.M.; Martínez-Reding, J.O. Immunohistochemical Diagnosis of Primary Cardiac Leiomyosarcoma in a Latin American Patient. Rare Tumors 2017, 9, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-G.; Li, N.-N. Primary Cardiac Synovial Sarcoma. Ann. Thorac. Surg. 2013, 95, 2202–2209. [Google Scholar] [CrossRef]
- Varma, T.; Adegboyega, P. Primary Cardiac Synovial Sarcoma. Arch. Pathol. Lab. Med. 2012, 136, 454–458. [Google Scholar] [CrossRef]
- Oizumi, S.; Igarashi, K.; Takenaka, T.; Yamashiro, K.; Hiraga, H.; Fujino, T.; Horimoto, M. Primary Pericardial Synovial Sarcoma With Detection of the Chimeric Transcript SYT-SSX. Jpn. Circ. J. 1999, 63, 330–332. [Google Scholar] [CrossRef] [Green Version]
- Casselman, F.P.; Gillinov, A.; Kasirajan, V.; Ratliff, N.B.; Cosgrove, D.M. Primary synovial sarcoma of the left heart. Ann. Thorac. Surg. 1999, 68, 2329–2331. [Google Scholar] [CrossRef]
- Paraf, F.; Bruneval, P.; Balaton, A.; Deloche, A.; Mikol, J.; Maitre, F.; Scholl, J.M.; De Saint-Maur, P.P.; Camilleri, J.P. Primary liposarcoma of the heart. Am. J. Cardiovasc. Pathol. 1990, 3, 175–180. [Google Scholar]
- Jakowski, J.D.; Wakely, P.E. Primary intrathoracic low-grade fibromyxoid sarcoma. Hum. Pathol. 2008, 39, 623–628. [Google Scholar] [CrossRef]
- Wang, J.-G.; Liu, B.; Gao, H.; Li, Y.-J.; Zhao, P.; Liu, X.-P. Primary Cardiac Osteosarcoma. Heart Lung Circ. 2016, 25, 698–704. [Google Scholar] [CrossRef]
- Burke, A.P.; Virmani, R. Osteosarcomas of the Heart. Am. J. Surg. Pathol. 1991, 15, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Hong, G.-R.; Youn, Y.-N.; Ha, J.-W.; Shim, C.Y. Primary cardiac chondrosarcoma mimicking chronic pulmonary thromboembolism in multimodality imaging. Eur. Hear. J. Cardiovasc. Imaging 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, X.; Guo, L.; Feng, Q.; Ni, Y. Primary Cardiac Chondrosarcoma. J. Card. Surg. 2012, 27, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-L.; Lee, D.; Chiou, K.-R.; Kung, M.-H.; Lin, S.-L.; Liu, C.-P.; Chiang, H.-T. Echocardiographic features of primary cardiac sarcoma. Echocardiography 2002, 19, 215–220. [Google Scholar] [CrossRef]
- Meng, Q.; Lai, H.; Lima, J.; Tong, W.; Qian, Y.; Lai, S. Echocardiographic and pathologic characteristics of primary cardiac tumors: A study of 149 cases. Int. J. Cardiol. 2002, 84, 69–75. [Google Scholar] [CrossRef]
- Kupsky, D.F.; Newman, D.B.; Kumar, G.; Maleszewski, J.J.; Edwards, W.D.; Klarich, K.W. Echocardiographic Features of Cardiac Angiosarcomas: The Mayo Clinic Experience (1976-2013). Echocardiography 2015, 33, 186–192. [Google Scholar] [CrossRef]
- Mankad, R.; Herrmann, J. Cardiac tumors: Echo assessment. Echo Res. Pract. 2016, 3, R65–R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohsaka, S.; Ara, V.; Feghali, S.F.; Stainback, R. Harmonic contrast transesophageal echocardiography of a soft-parts sarcoma metastatic to the heart. Tex. Hear. Inst. J. 2005, 32, 610–611. [Google Scholar]
- Lang, R.M.; Mor-Avi, V. Clinical utility of contrast-enhanced echocardiography. Clin. Cardiol. 2006, 29, I15–I25. [Google Scholar] [CrossRef]
- Kirkpatrick, J.N.; Wong, T.; Bednarz, J.E.; Spencer, K.T.; Sugeng, L.; Ward, R.; DeCara, J.M.; Weinert, L.; Krausz, T.; Lang, R.M. Differential diagnosis of cardiac masses using contrast echocardiographic perfusion imaging. J. Am. Coll. Cardiol. 2004, 43, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Guta, A.C.; Badano, L.P.; Ochoa-Jimenez, R.C.; Genovese, D.; Previtero, M.; Civera, S.; Ruocco, A.; Bettella, N.; Parati, G.; Muraru, D. Three-dimensional echocardiography to assess left ventricular geometry and function. Expert Rev. Cardiovasc. Ther. 2019, 17, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Asch, F.M.; Bieganski, S.P.; Panza, J.A.; Weissman, N.J. Real-Time 3-Dimensional Echocardiography Evaluation of Intracardiac Masses. Echocardiography 2006, 23, 218–224. [Google Scholar] [CrossRef]
- Gok, G.; Elsayed, M.; Thind, M.; Uygur, B.; Abtahi, F.; Chahwala, J.R.; Yıldırımtürk, Ö.; Kayacıoğlu, İ.; Pehlivanoğlu, S.; Nanda, N.C. Incremental value of live/real time three-dimensional transesophageal echocardiography over the two-dimensional technique in the assessment of primary cardiac malignant fibrous histiocytoma. Echocardiography 2015, 32, 1164–1170. [Google Scholar] [CrossRef]
- Reddy, V.K.; Faulkner, M.; Bandarupalli, N.; Nanda, N.C.; Singh, P.; Dutta, R.; Singh, A.; Pothineni, K.R.; Dod, H.S.; Bhardwaj, R.; et al. Incremental Value of Live/Real Time Three-Dimensional Transthoracic Echocardiography in the Assessment of Right Ventricular Masses. Echocardiography 2009, 26, 598–609. [Google Scholar] [CrossRef]
- Hoey, E.T.; Shahid, M.; Ganeshan, A.; Baijal, S.; Simpson, H.; Watkin, R.W. MRI assessment of cardiac tumours: Part 1, multiparametric imaging protocols and spectrum of appearances of histologically benign lesions. Quant. Imaging Med. Surg. 2014, 4, 478–488. [Google Scholar] [CrossRef]
- Hoey, E.T.; Shahid, M.; Ganeshan, A.; Baijal, S.; Simpson, H.; Watkin, R.W. MRI assessment of cardiac tumours: Part 2, spectrum of appearances of histologically malignant lesions and tumour mimics. Quant. Imaging Med. Surg. 2014, 4, 489–497. [Google Scholar] [CrossRef]
- Cazalbou, S.; Shen, V.C.F.; Petermann, A.; Eyharts, D.; Fournier, P.; Cariou, E.; Lavie-Badie, Y.; Hennig, A.; Roncalli, J.; Rousseau, H.; et al. What is the most useful imaging parameter to explore the prognostic value of the right ventricular function at the time of multimodality cardiovascular imaging? Echocardiography 2020, 37, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, N.; Cheezum, M.K.; Aghayev, A.; Padera, R.; Vita, T.; Steigner, M.; Hulten, E.; Bittencourt, M.S.; Dorbala, S.; Di Carli, M.F.; et al. Assessment of Cardiac Masses by Cardiac Magnetic Resonance Imaging: Histological Correlation and Clinical Outcomes. J. Am. Hear. Assoc. 2019, 8, e007829. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.T.; Fox, J.; Perez Johnston, R.; Kim, J.; Brouwer, L.R.; Grizzard, J.; Kim, R.J.; Matasar, M.; Shia, J.; Moskowitz, C.S.; et al. Late Gadolinium Enhancement Cardiac Magnetic Resonance Tissue Characterization for Cancer-Associated Cardiac Masses: Metabolic and Prognostic Manifestations in Relation to Whole-Body Positron Emission Tomography. J. Am. Heart Assoc. 2019, 8, e011709. [Google Scholar] [CrossRef] [Green Version]
- Araoz, P.A.; Eklund, H.E.; Welch, T.J.; Breen, J.F. CT and MR Imaging of Primary Cardiac Malignancies. Radiographics 1999, 19, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, A.M.; Mariki, D.E.; Gundah, C.M.; Assey, E.V.; Zwetselaar, M.; Howlett, W.P.; Dekker, M.C.J. Feeling the price tag of magnetic resonance imaging claustrophobia. J. Magn. Reson. Imaging 2021, 54, 328. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Luo, R.; Wei, Y.; Wang, F.; Zhang, Y.; Karlson, K.J.; Zhang, Z.; Reardon, M.J.; Dobrilovic, N. Survival outcomes in patients with primary cardiac sarcoma in the United States. J. Thorac. Cardiovasc. Surg. 2021, 162, 107–115.e2. [Google Scholar] [CrossRef] [PubMed]
- Mkalaluh, S.; Szczechowicz, M.; Torabi, S.; Schmack, B.; Sabashnikov, A.; Dib, B.; Karck, M.; Weymann, A. Surgical Treatment of Cardiac Tumors: Insights from an 18-Year Single-Center Analysis. Med. Sci. Monit. 2017, 23, 6201–6209. [Google Scholar] [CrossRef] [Green Version]
- Reardon, M.J.; Malaisrie, S.C.; Walkes, J.-C.; Vaporciyan, A.A.; Rice, D.C.; Smythe, W.R.; DeFelice, C.A.; Wojciechowski, Z.J. Cardiac Autotransplantation for Primary Cardiac Tumors. Ann. Thorac. Surg. 2006, 82, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Blackmon, S.H.; Patel, A.R.; Bruckner, B.A.; Beyer, E.A.; Rice, D.C.; Vaporciyan, A.A.; Wojciechowski, Z.; Correa, A.M.; Reardon, M.J. Cardiac Autotransplantation for Malignant or Complex Primary Left-Heart Tumors. Tex. Heart Inst. J. 2008, 35, 296–300. [Google Scholar] [PubMed]
- Reardon, M.J.; DeFelice, C.A.; Sheinbaum, R.; Baldwin, J.C. Cardiac autotransplant for surgical treatment of a malignant neoplasm. Ann. Thorac. Surg. 1999, 67, 1793–1795. [Google Scholar] [CrossRef]
- Gowdamarajan, A.; Michler, R.E. Therapy for primary cardiac tumors: Is there a role for heart transplantation? Curr. Opin. Cardiol. 2000, 15, 121–125. [Google Scholar] [CrossRef]
- Li, H.; Yang, S.; Chen, H.; Yang, Z.; Hong, T.; Hou, Y.; Wang, C. Survival after heart transplantation for non-metastatic primary cardiac sarcoma. J. Cardiothorac. Surg. 2016, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Correa, A.M.; Blackmon, S.; Quiroga-Garza, G.; Weilbaecher, D.; Bruckner, B.; Ramlawi, B.; Rice, D.C.; Vaporciyan, A.A.; Reardon, M.J. Outcomes After Right-Side Heart Sarcoma Resection. Ann. Thorac. Surg. 2011, 91, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ramlawi, B.; Leja, M.J.; Abu Saleh, W.K.; Al Jabbari, O.; Benjamin, R.; Ravi, V.; Shapira, O.M.; Blackmon, S.H.; Bruckner, B.A.; Reardon, M.J. Surgical Treatment of Primary Cardiac Sarcomas: Review of a Single-Institution Experience. Ann. Thorac. Surg. 2016, 101, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Xu, D.; Chen, Z.; Ding, W.; Hong, T.; Chen, H.; Shao, M.; Lai, H.; Hou, Y.; Wang, C. Prognostic Analysis for Survival After Resections of Localized Primary Cardiac Sarcomas: A Single-Institution Experience. Ann. Thorac. Surg. 2014, 97, 1379–1385. [Google Scholar] [CrossRef]
- Agaimy, A.; Rösch, J.; Weyand, M.; Strecker, T. Primary and metastatic cardiac sarcomas: A 12-year experience at a German heart center. Int. J. Clin. Exp. Pathol. 2012, 5, 928–938. [Google Scholar]
- Madershahian, N.; Reuter, H.; Bangard, C.; Baldus, S.; Wahlers, T.; Wippermann, J.; Deppe, A.-C.; Adler, C. Cardiac Liposarcoma—A Review of Outcome after Surgical Resection. Thorac. Cardiovasc. Surg. 2013, 62, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Clément-Colmou, K.; Vogin, G.; Beckendorf, V.; Ducassou, A.; Ali, A.M.; Salas, S.; Saada, E.; Thyss, A.; Lapeyre, M.; et al. Radiation therapy of cardiac sarcomas. Cancer Radiother. 2014, 18, 125–131. [Google Scholar] [CrossRef]
- Fatima, J.; Duncan, A.A.; Maleszewski, J.J.; Kalra, M.; Oderich, G.S.; Gloviczki, P.; Suri, R.M.; Bower, T.C. Primary angiosarcoma of the aorta, great vessels, and the heart. J. Vasc. Surg. 2013, 57, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Huang, C.; Zhang, X.; Zhao, L.; Pan, D. Clinical characteristics associated with primary cardiac angiosarcoma outcomes: A surveillance, epidemiology and end result analysis. Eur. J. Med. Res. 2019, 24, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Aboud, A.; Farha, K.; Hsieh, W.C.; Brasch, F.; Ensminger, S.; Gummert, J.; Renner, A. Prognostic Factors for Long-Term Survival after Surgical Resection of Primary Cardiac Sarcoma. Thorac. Cardiovasc. Surg. 2019, 67, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Elsayad, K.; Lehrich, P.; Yppaerilae-Wolters, H.; Dieckmann, C.; Kriz, J.; Haverkamp, U.; Eich, H.T.; Information, P.E.K.F.C. Primary Cardiac Angiosarcoma Treated With Positron Emission Tomography/Magnetic Resonance Imaging–Guided Adaptive Radiotherapy. Can. J. Cardiol. 2016, 32, 829.e7–829.e10. [Google Scholar] [CrossRef] [PubMed]
- Moeri-Schimmel, R.; Pras, E.; Desar, I.; Krol, S.; Braam, P. Primary sarcoma of the heart: Case report and literature review. J. Cardiothorac. Surg. 2020, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hong, N.J.L.; Pandalai, P.; Hornick, J.; Shekar, P.S.; Harmon, D.C.; Chen, Y.-L.; Butrynski, J.E.; Baldini, E.H.; Raut, C.P. Cardiac Angiosarcoma Management and Outcomes: 20-Year Single-institution Experience. Ann. Surg. Oncol. 2012, 19, 2707–2715. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Million, L.; Moding, E.J.; Scott, G.; Berry, M.; Ganjoo, K.N. The impact of postoperative therapy on primary cardiac sarcoma. J. Thorac. Cardiovasc. Surg. 2018, 156, 2194–2203. [Google Scholar] [CrossRef]
- Wang, M.; Fu, G.; Jiang, H.; Zeng, K.; Hua, P. Multimodality Treatment for Cardiac Angiosarcoma. Intern. Med. 2014, 53, 1949–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Saleh, W.K.; Ramlawi, B.; Shapira, O.M.; Al Jabbari, O.; Ravi, V.; Benjamin, R.; Durand, J.-B.; Leja, M.J.; Blackmon, S.H.; Bruckner, B.A.; et al. Improved Outcomes With the Evolution of a Neoadjuvant Chemotherapy Approach to Right Heart Sarcoma. Ann. Thorac. Surg. 2017, 104, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Heinhuis, K.M.; Ijzerman, N.S.; Van Der Graaf, W.T.A.; Kerst, J.M.; Schrage, Y.; Beijnen, J.H.; Steeghs, N.; Van Houdt, W.J. Neoadjuvant Systemic Treatment of Primary Angiosarcoma. Cancers 2020, 12, 2251. [Google Scholar] [CrossRef]
- Ravi, V.; Benjamin, R.S. Systemic therapy for cardiac sarcomas. Methodist DeBakey Cardiovasc. J. 2010, 6, 57–60. [Google Scholar] [CrossRef]
- Frezza, A.M.; Assi, T.; Vullo, S.L.; Ben-Ami, E.; Dufresne, A.; Yonemori, K.; Noguchi, E.; Siontis, B.; Ferraro, R.; Teterycz, P.; et al. Systemic treatments in MDM2 positive intimal sarcoma: A multicentre experience with anthracycline, gemcitabine, and pazopanib within the World Sarcoma Network. Cancer 2020, 126, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Singhi, E.K.; Moore, D.C.; Muslimani, A. Metastatic Soft Tissue Sarcomas: A Review of Treatment and New Pharmacotherapies. J. Formul. Manag. 2018, 43, 410–429. [Google Scholar]
- Demetri, G.D.; Von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Sultan, I.; Bianco, V.; Habertheuer, A.; Kilic, A.; Gleason, T.G.; Aranda-Michel, E.; Harinstein, M.E.; Martinez-Meehan, D.; Arnaoutakis, G.; Okusanya, O. Long-Term Outcomes of Primary Cardiac Malignancies: Multi-Institutional Results From the National Cancer Database. J. Am. Coll. Cardiol. 2020, 75, 2338–2347. [Google Scholar] [CrossRef]
- Llombart-Cussac, A.; Pivot, X.; Contesso, G.; Rhor-Alvarado, A.; Delord, J.P.; Spielmann, M.; Türsz, T.; Le Cesne, A. Adjuvant chemotherapy for primary cardiac sarcomas: The IGR experience. Br. J. Cancer 1998, 78, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, B.S.; Stahl, K.A.; Hollenbeak, C.S.; Taylor, M.D.; Vasekar, M.K.; Drabick, J.J.; Conte, J.V.; Soleimani, B.; Reed, M.F. Postoperative chemotherapy and radiation improve survival following cardiac sarcoma resection. J. Thorac. Cardiovasc. Surg. 2021, 161, 110–119.e4. [Google Scholar] [CrossRef]
- Zhrebker, L.; Cherni, I.; Gross, L.M.; Hinshelwood, M.M.; Reese, M.; Aldrich, J.; Guileyardo, J.M.; Roberts, W.C.; Craig, D.; Von Hoff, D.D.; et al. Case report: Whole exome sequencing of primary cardiac angiosarcoma highlights potential for targeted therapies. BMC Cancer 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Mehta, C.; Liu, L.; Theuer, C. An adaptive population enrichment phase III trial of TRC105 and pazopanib versus pazopanib alone in patients with advanced angiosarcoma (TAPPAS trial). Ann. Oncol. 2019, 30, 103–108. [Google Scholar] [CrossRef]
- Boekstegers, P.; Von Degenfeld, G.; Giehrl, W.; Heinrich, D.; Hullin, R.; Kupatt, C.; Steinbeck, G.; Baretton, G.; Middeler, G.; Katus, H.; et al. Myocardial gene transfer by selective pressure-regulated retroinfusion of coronary veins. Gene Ther. 2000, 7, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Vesely, D.L. Cardiac hormones for the treatment of cancer. Endocr. Relat. Cancer 2013, 20, R113–R125. [Google Scholar] [CrossRef] [Green Version]
- Siozopoulou, V.; Domen, A.; Zwaenepoel, K.; Van Beeck, A.; Smits, E.; Pauwels, P.; Marcq, E. Immune Checkpoint Inhibitory Therapy in Sarcomas: Is There Light at the End of the Tunnel? Cancers 2021, 13, 360. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Rosenberg, A.E.; Wieder, E.; Komanduri, K.V.; Kolonias, D.; Uduman, M.; Castle, J.C.; Buell, J.S.; Trent, J.C.; Wilky, B.A. Angiosarcoma patients treated with immune checkpoint inhibitors: A case series of seven patients from a single institution. J. Immunother. Cancer 2019, 7, 213. [Google Scholar] [CrossRef] [Green Version]
- Le Cesne, A.; Marec-Berard, P.; Blay, J.-Y.; Gaspar, N.; Bertucci, F.; Penel, N.; Bompas, E.; Cousin, S.; Toulmonde, M.; Bessede, A.; et al. Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: Results from the PEMBROSARC study. Eur. J. Cancer 2019, 119, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2019, 8, e000798. [Google Scholar] [CrossRef] [PubMed]
- Birdi, H.K.; Jirovec, A.; Cortés-Kaplan, S.; Werier, J.; Nessim, C.; Diallo, J.-S.; Ardolino, M. Immunotherapy for sarcomas: New frontiers and unveiled opportunities. J. Immunother. Cancer 2021, 9, e001580. [Google Scholar] [CrossRef]
- Bonomo, P.; Cipressi, S.; Desideri, I.; Masi, L.; Doro, R.; Iermano, C.; Greto, D.; Simontacchi, G.; Mangoni, M.; Paiar, F.; et al. Stereotactic Body Radiotherapy with Cyberknife for Cardiac Malignancies. Tumori J. 2015, 101, 294–297. [Google Scholar] [CrossRef]
- Jacobs, S.; Grunert, R.; Mohr, F.W.; Falk, V. 3D-Imaging of cardiac structures using 3D heart models for planning in heart surgery: A preliminary study. Interact. Cardiovasc. Thorac. Surg. 2008, 7, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Sim, A.J.; Palm, R.F.; DeLozier, K.B.; Feygelman, V.; Latifi, K.; Redler, G.; Washington, I.R.; Wuthrick, E.J.; Rosenberg, S.A. MR-guided stereotactic body radiation therapy for intracardiac and pericardial metastases. Clin. Transl. Radiat. Oncol. 2020, 25, 102–106. [Google Scholar] [CrossRef]
- Saponara, M.; Indio, V.; Pizzi, C.; Serban, E.-D.; Urbini, M.; Astolfi, A.; Paolisso, P.; Suarez, S.M.; Nannini, M.; Pacini, D.; et al. Successful multidisciplinary clinical approach and molecular characterization by whole transcriptome sequencing of a cardiac myxofibrosarcoma: A case report. World J. Clin. Cases 2019, 7, 3018–3026. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Sarcoma | AFIP Registry Period: <1974–1993 | JCS Registry Period: 1999–2010 | JATS Registry Period: 1999–2010 | French Sarcoma Group Period: 1977–2010 % among PCSs |
|---|---|---|---|---|
| Angiosarcoma | 7.3–8.5% | 9.5% | 8.2% | 32.3% |
| Undifferentiated high grade pleomorphic sarcoma | 4.1–8.5%% | 3.3% | 4.3% | 36.3% |
| Leiomyosarcoma | 0.19–3.1% | 1.9% | 2.2% | 12.9% |
| Rhabdomyosarcoma | 1.6–4.9% | 0.8% | 0.45% | 18.6% |
| Synovial sarcoma | 0.19–1.4% | 0.8% | 0.9% | |
| Liposarcoma | 0.19–0.5% | 1.1% | 0.9% | |
| Fibrosarcoma and myxoid fibrosarcoma | 2.3–3.2% | 0.3% | 0% | |
| Osteosarcoma/Chondrosarcoma | 0.94–4.7% | 0–0.8% | 0.45–1.3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scicchitano, P.; Sergi, M.C.; Cameli, M.; Miglioranza, M.H.; Ciccone, M.M.; Gentile, M.; Porta, C.; Tucci, M. Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology. Biomedicines 2021, 9, 774. https://doi.org/10.3390/biomedicines9070774
Scicchitano P, Sergi MC, Cameli M, Miglioranza MH, Ciccone MM, Gentile M, Porta C, Tucci M. Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology. Biomedicines. 2021; 9(7):774. https://doi.org/10.3390/biomedicines9070774
Chicago/Turabian StyleScicchitano, Pietro, Maria Chiara Sergi, Matteo Cameli, Marcelo H. Miglioranza, Marco Matteo Ciccone, Marica Gentile, Camillo Porta, and Marco Tucci. 2021. "Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology" Biomedicines 9, no. 7: 774. https://doi.org/10.3390/biomedicines9070774
APA StyleScicchitano, P., Sergi, M. C., Cameli, M., Miglioranza, M. H., Ciccone, M. M., Gentile, M., Porta, C., & Tucci, M. (2021). Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology. Biomedicines, 9(7), 774. https://doi.org/10.3390/biomedicines9070774