
LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis

 ,
,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation of Specific Brain and Spinal Cord Regions
2.3. Cell Culture
2.4. RNA Extraction
2.5. Real-Time PCR
3. Results
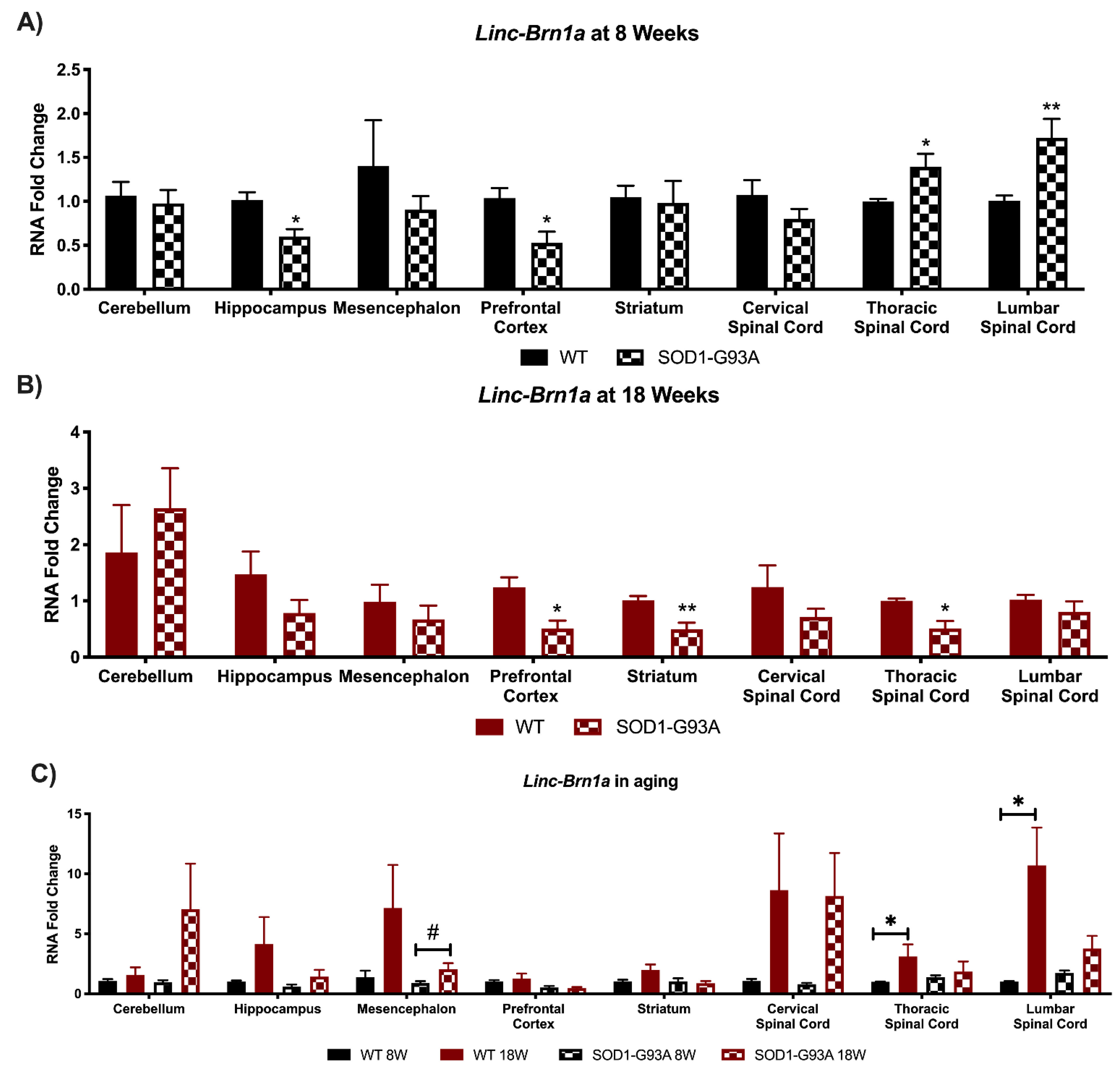
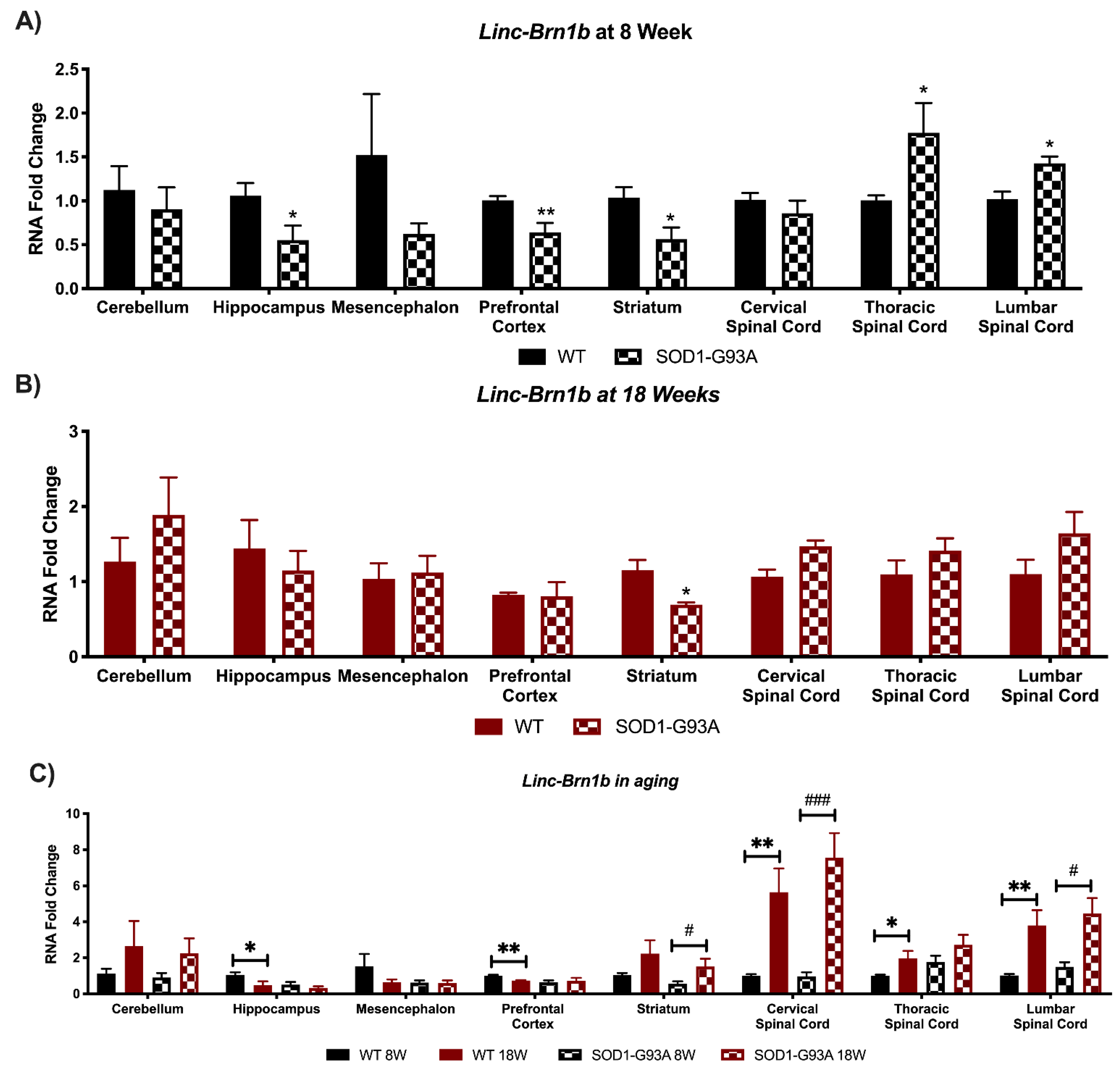
3.1. Linc-Brn1a and Linc-Brn1b Present Altered Expression in CNS Areas of SOD1-G93A Mice
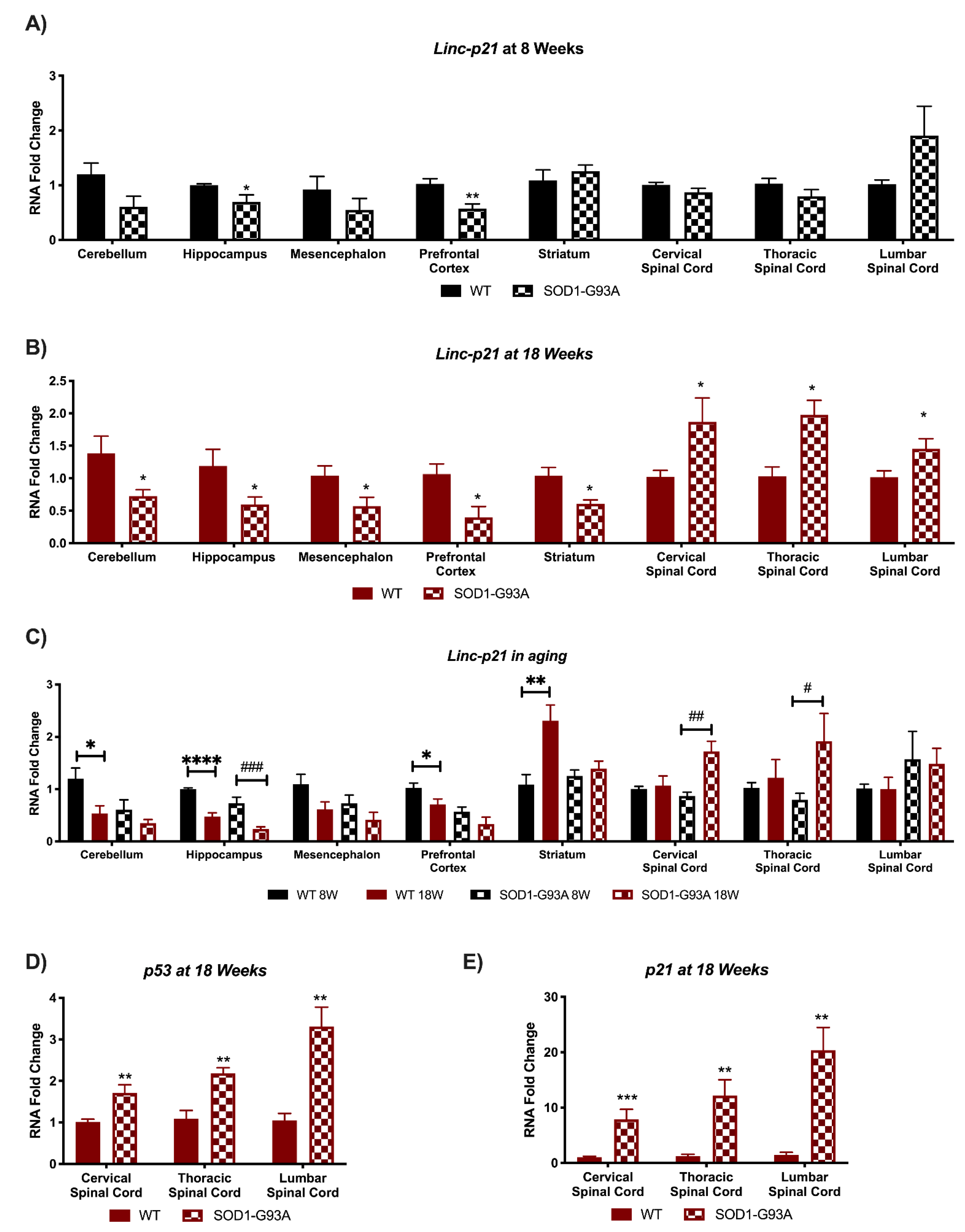
3.2. Linc-p21 Presents with Altered Expression in All CNS Areas of Symptomatic SOD1-G93A Mice
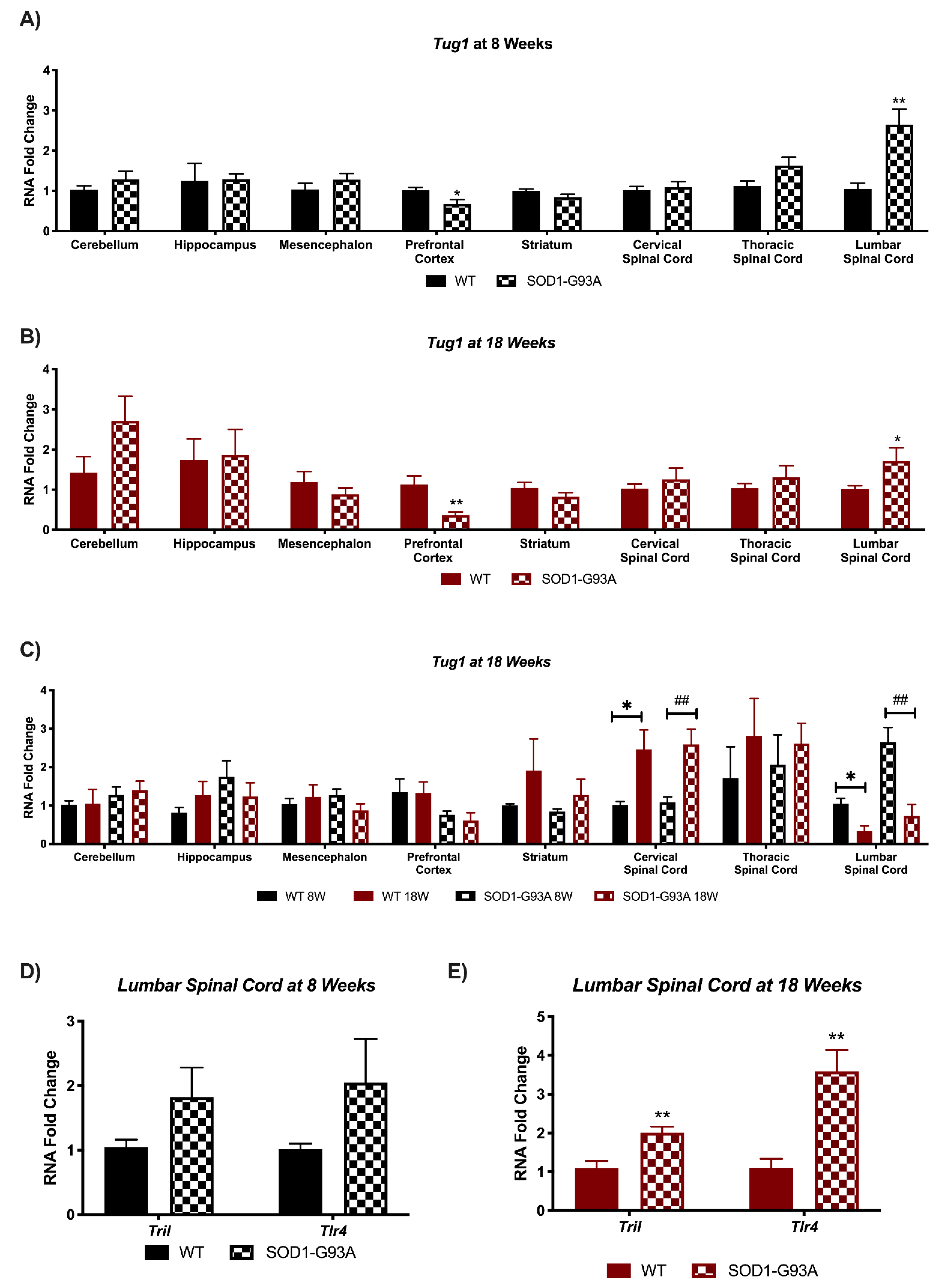
3.3. Tug1 Expression Is Deregulated in Prefrontal Cortex and Lumbar Spinal Cord of Pre- and Symptomatic SOD1-G93A Mice
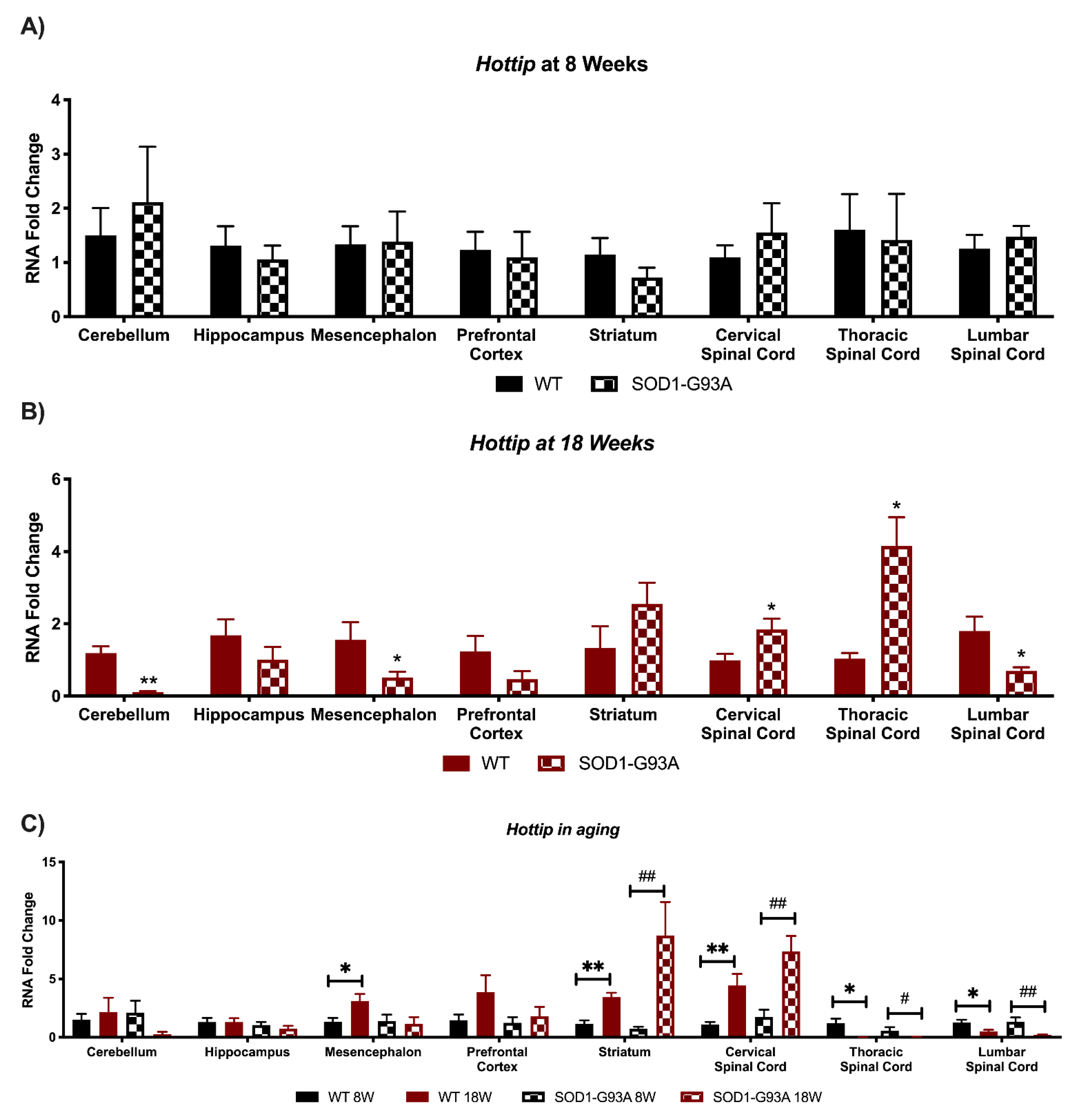
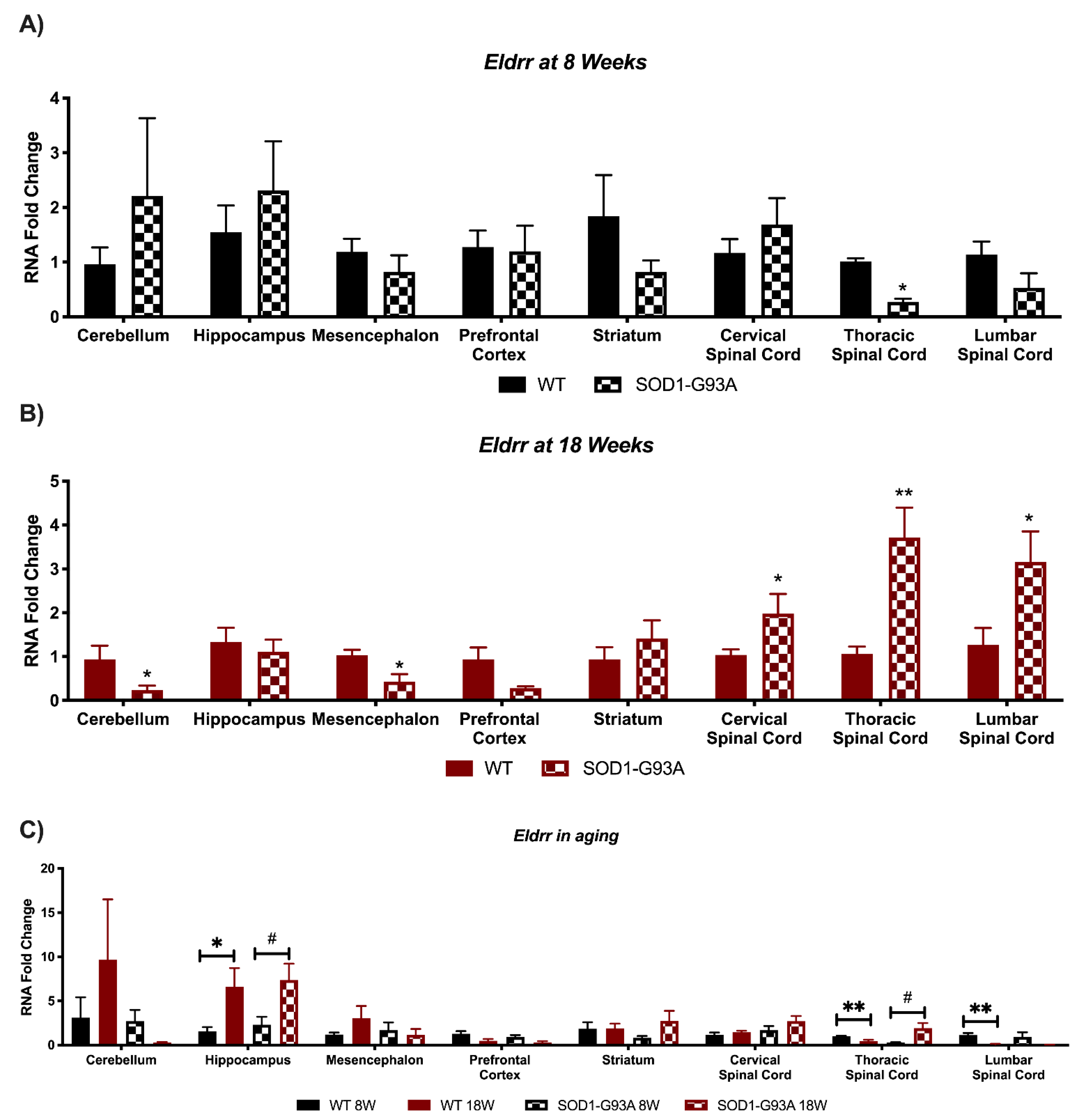
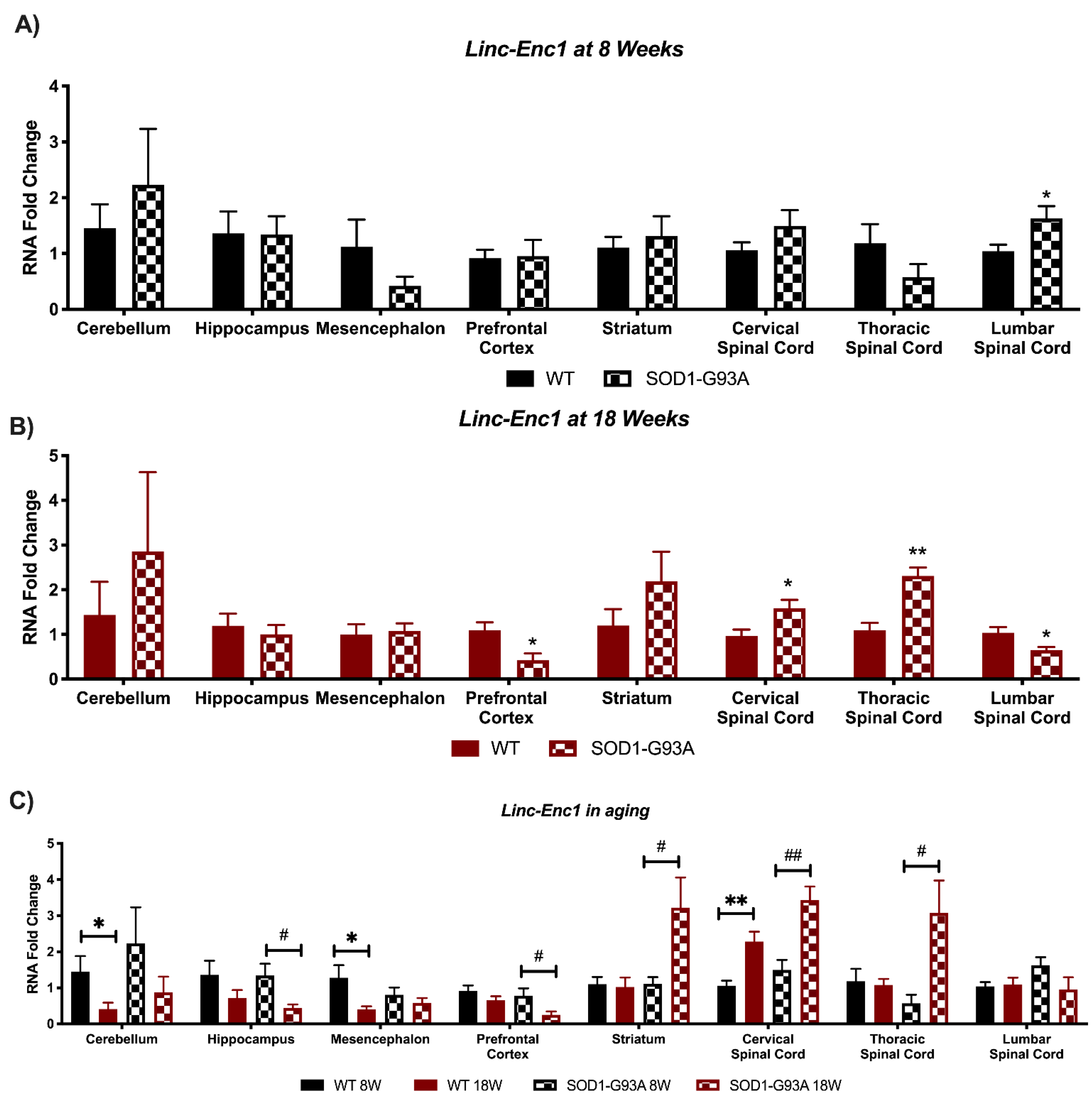
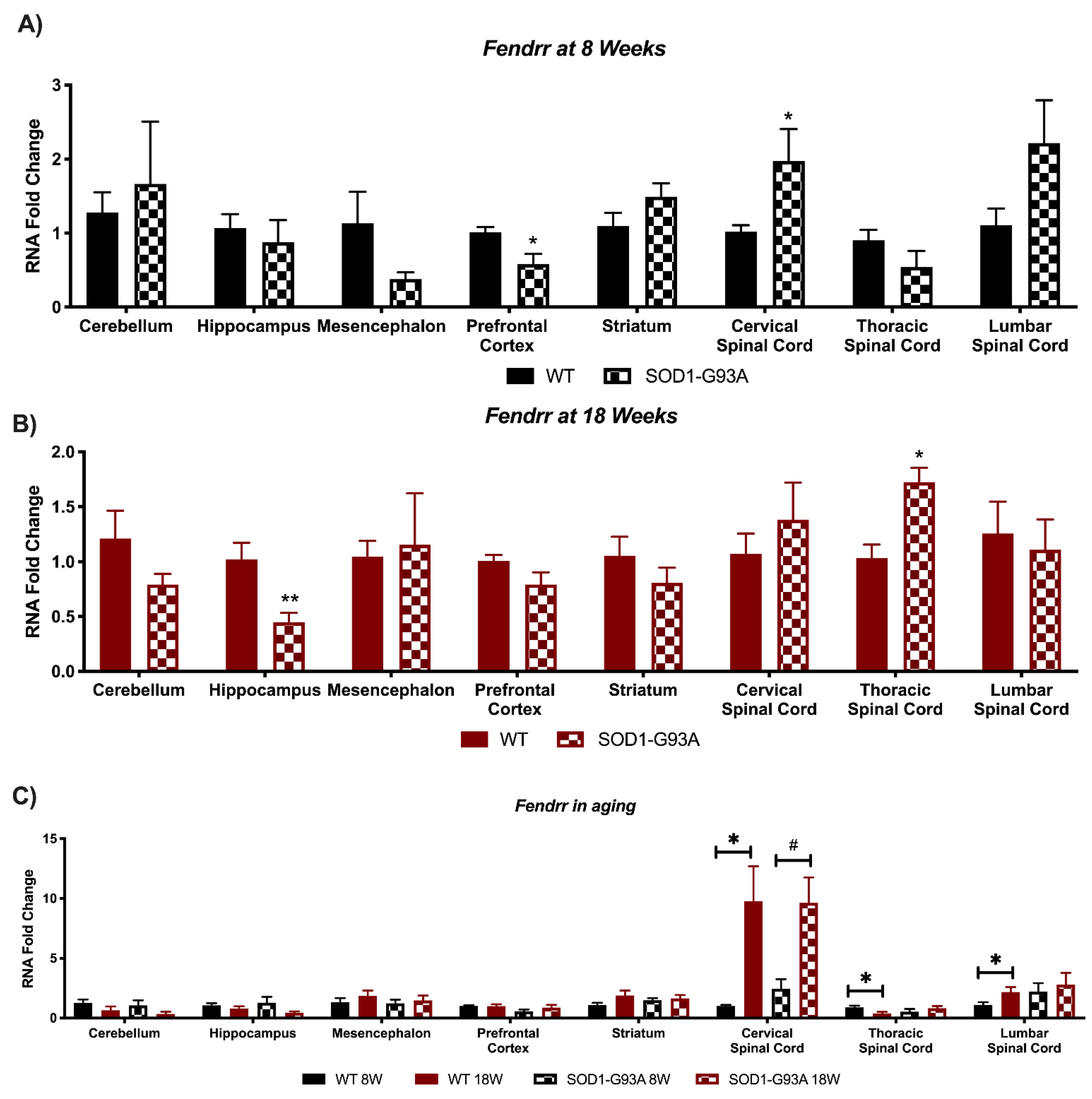
3.4. Expression Analysis of Hottip, Eldrr, Linc-Enc1, Fendrr Reveals Global Perturbation in SOD1-G93A Mice
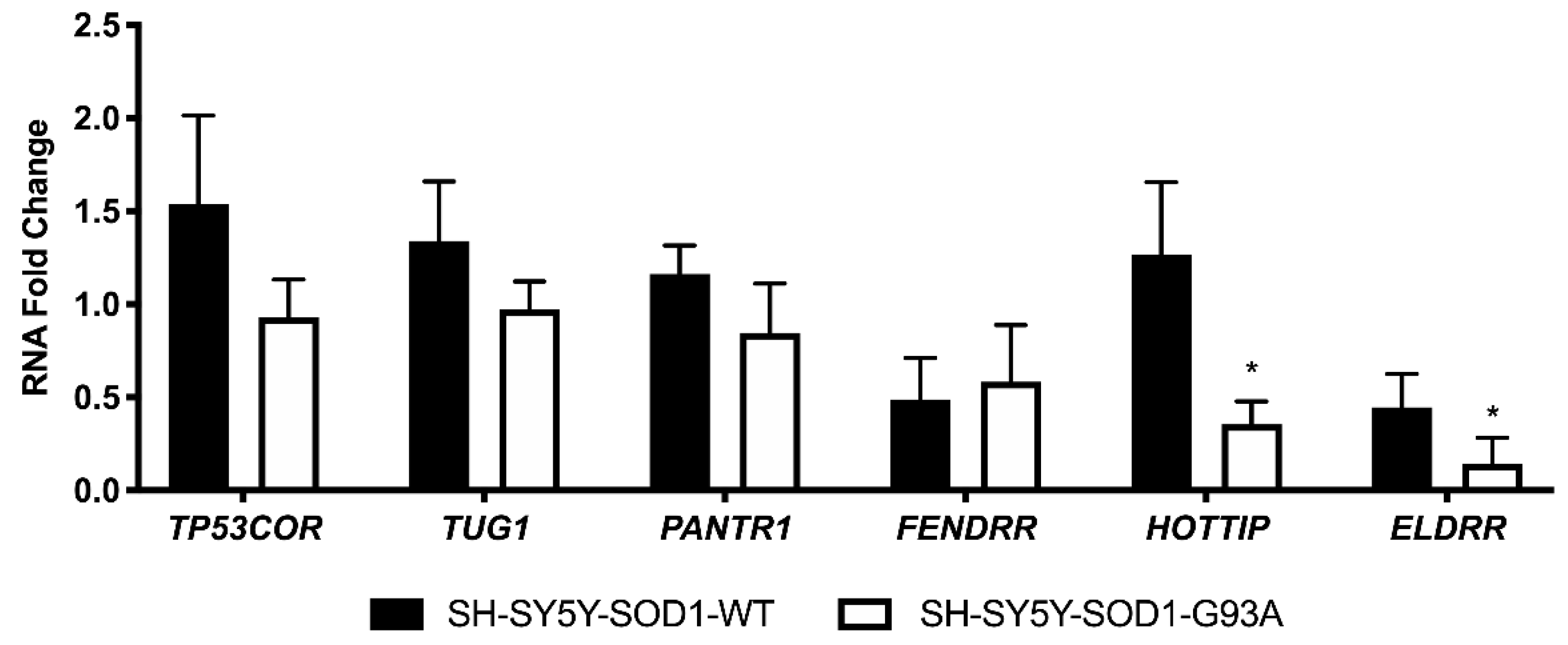
3.5. The Expression of Human Homologues of Investigated lncRNAs in an In Vitro Model of ALS Resulted Significantly Deregulated
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijesekera, L.L.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Hentati, A.; Tainer, J.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Pandini, C.; Garofalo, M.; Bordoni, M.; Pansarasa, O.; Cereda, C. Long non coding RNAs and ALS: Still much to do. Non-Coding RNA Res. 2018, 3, 226–231. [Google Scholar] [CrossRef]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Model. Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Bonanno, S.; Kapetis, D.; Barzago, C.; Cavalcante, P.; D’Alessandro, S.; Mantegazza, R.; Bernasconi, P. Up-regulation of neural and cell cycle-related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Mol. Brain 2015, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzo, S.; Kapetis, D.; Mantegazza, R.; Baggi, F.; Bonanno, S.; Barzago, C.; Cavalcante, P.; de Rosbo, N.K.; Bernasconi, P. Altered miRNA expression is associated with neuronal fate in G93A-SOD1 ependymal stem progenitor cells. Exp. Neurol. 2014, 253, 91–101. [Google Scholar] [CrossRef]
- Alrafiah, A.R. From Mouse Models to Human Disease: An Approach for Amyotrophic Lateral Sclerosis. In Vivo 2018, 32, 983–998. [Google Scholar] [CrossRef] [Green Version]
- Galán, L.; Pinedo, U.; Vela-Souto, A.; Guerrero-Sola, A.; Barcia, J.A.; Gutierrez, A.R.; Martinez-Martinez, A.; Jiménez, M.S.B.; García-Verdugo, J.M.; Matías-Guiu, J. Subventricular zone in motor neuron disease with frontotemporal dementia. Neurosci. Lett. 2011, 499, 9–13. [Google Scholar] [CrossRef]
- Sauvageau, M.; Goff, L.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2013, 2, e01749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, S.; Giallongo, T.; Rey, F.; Latorre, E.; Bordoni, M.; Mazzucchelli, S.; Gorio, M.C.; Pansarasa, O.; Provenzani, A.; Cereda, C.; et al. HuR interacts with lincBRN1a and lincBRN1b during neuronal stem cells differentiation. RNA Biol. 2019, 16, 1471–1485. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2019, 11, 77–89. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, H.-L.; Hu, G.-W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.-M.; Wu, L.; Xu, G.-H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar] [PubMed]
- Li, W.; Wu, X.; She, W. LncRNA POU3F3 promotes cancer cell migration and invasion in nasopharyngeal carcinoma by up-regulating TGF-β1. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Wang, K.; Yeh, S.; Sun, Y.; Liang, L.; Xiao, Y.; Xu, W.; Niu, Y.; Cheng, L.; Maity, S.N.; et al. LncRNA-p21 alters the antiandrogen enzalutamide-induced prostate cancer neuroendocrine differentiation via modulating the EZH2/STAT3 signaling. Nat. Commun. 2019, 10, 2571. [Google Scholar] [CrossRef] [Green Version]
- Ao, X.; Jiang, M.; Zhou, J.; Liang, H.; Xia, H.; Chen, G. lincRNA-p21 inhibits the progression of non-small cell lung cancer via targeting miR-17-5p. Oncol. Rep. 2018, 41, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Kosari-Monfared, M.; Golpour, M.; Emami, Z.; Ghasemiyan, M.; Nouri, M.; Akhavan-Niaki, H. LncRNAs as potential diagnostic and prognostic biomarkers in gastric cancer: A novel approach to personalized medicine. J. Cell. Physiol. 2020, 235, 3189–3206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hu, T.; Xu, Y. Anticancer potential of TUG1 knockdown in cisplatin-resistant osteosarcoma through inhibition of MET/Akt signalling. J. Drug Target. 2019, 28, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, Z.; Song, E.; Hu, P.; Yang, Q.; Hu, Y.; Liu, H.; Jin, A. LncRNA HOTTIP enhances human osteogenic BMSCs differentiation via interaction with WDR5 and activation of Wnt/β-catenin signalling pathway. Biochem. Biophys. Res. Commun. 2020, 524, 1037–1043. [Google Scholar] [CrossRef]
- Xiong, L.; Tang, Y.; Tang, J.; Liu, Z.; Wang, X. Downregulation of lncRNA HOTTIP Suppresses the Proliferation, Migration, and Invasion of Oral Tongue Squamous Cell Carcinoma by Regulation of HMGA2-Mediated Wnt/β-Catenin Pathway. Cancer Biother. Radiopharm. 2020, 35, 720–730. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhao, H. Identification of a nomogram based on long non-coding RNA to improve prognosis prediction of esophageal squamous cell carcinoma. Aging 2020, 12, 1512–1526. [Google Scholar] [CrossRef]
- Xu, X.; Zhuang, C.; Wu, Z.; Qiu, H.; Feng, H.; Wu, J. LincRNA-p21 Inhibits Cell Viability and Promotes Cell Apoptosis in Parkinson’s Disease through Activating α-Synuclein Expression. BioMed Res. Int. 2018, 2018, 8181374. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: A longitudinal MRI study. Exp. Neurol. 2011, 231, 30–37. [Google Scholar] [CrossRef]
- Cova, E.; Ghiroldi, A.; Guareschi, S.; Mazzini, G.; Gagliardi, S.; Davin, A.; Bianchi, M.; Ceroni, M.; Cereda, C. G93A SOD1 alters cell cycle in a cellular model of Amyotrophic Lateral Sclerosis. Cell. Signal. 2010, 22, 1477–1484. [Google Scholar] [CrossRef]
- Goff, L.A.; Groff, A.F.; Sauvageau, M.; Trayes-Gibson, Z.; Sanchez-Gomez, D.B.; Morse, M.; Martin, R.D.; Elcavage, L.; Liapis, S.C.; Gonzalez-Celeiro, M.; et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2015, 112, 6855–6862. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.; Gorospe, M. LincRNA-p21 Suppresses Target mRNA Translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A Large Intergenic Noncoding RNA Induced by p53 Mediates Global Gene Repression in the p53 Response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Ma, H.; Li, Z.; Chen, F.; Fang, B.; Cao, X.; Chang, Y.; Qiang, Z. Downregulation of LncRNA TUG1 Inhibited TLR4 Signaling Pathway-Mediated Inflammatory Damage After Spinal Cord Ischemia Reperfusion in Rats via Suppressing TRIL Expression. J. Neuropathol. Exp. Neurol. 2019, 78, 268–282. [Google Scholar] [CrossRef] [PubMed]
- A Pandya, V.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Hunter, T.A.; Maxwell, N.; Pereira, K.; Whittaker, C.A.; Valdez, G.; Guarente, L.P. SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 2018, 17, e12839. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Thorns, J.; Jansma, H.; Peschel, T.; Grosskreutz, J.; Mohammadi, B.; Dengler, R.; Münte, T.F. Extent of cortical involvement in amyotrophic lateral sclerosis – an analysis based on cortical thickness. BMC Neurol. 2013, 13, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán, L.; Gómez-Pinedo, U.; Guerrero, A.; García-Verdugo, J.M.; Matías-Guiu, J. Amyotrophic lateral sclerosis modifies progenitor neural proliferation in adult classic neurogenic brain niches. BMC Neurol. 2017, 17, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Schuster, J.E.; Fu, R.; Siddique, T.; Heckman, C. Progressive Changes in Synaptic Inputs to Motoneurons in Adult Sacral Spinal Cord of a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2009, 29, 15031–15038. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, F.; Marcuzzo, S.; Bonanno, S.; Bordoni, M.; Giallongo, T.; Malacarne, C.; Cereda, C.; Zuccotti, G.V.; Carelli, S. LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 809. https://doi.org/10.3390/biomedicines9070809
Rey F, Marcuzzo S, Bonanno S, Bordoni M, Giallongo T, Malacarne C, Cereda C, Zuccotti GV, Carelli S. LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis. Biomedicines. 2021; 9(7):809. https://doi.org/10.3390/biomedicines9070809
Chicago/Turabian StyleRey, Federica, Stefania Marcuzzo, Silvia Bonanno, Matteo Bordoni, Toniella Giallongo, Claudia Malacarne, Cristina Cereda, Gian Vincenzo Zuccotti, and Stephana Carelli. 2021. "LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis" Biomedicines 9, no. 7: 809. https://doi.org/10.3390/biomedicines9070809
APA StyleRey, F., Marcuzzo, S., Bonanno, S., Bordoni, M., Giallongo, T., Malacarne, C., Cereda, C., Zuccotti, G. V., & Carelli, S. (2021). LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis. Biomedicines, 9(7), 809. https://doi.org/10.3390/biomedicines9070809