
RGS4-Deficiency Alters Intracellular Calcium and PKA-Mediated Control of Insulin Secretion in Glucose-Stimulated Beta Islets

 , ,
, ,
Abstract
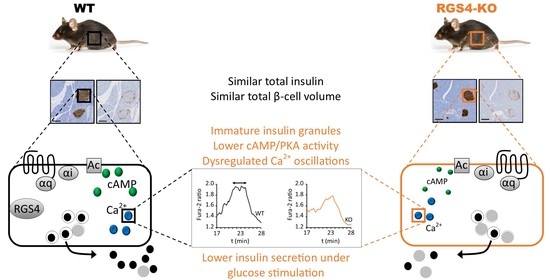
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Material
2.2. OGTT
2.3. Isolation and Culture of Pancreatic Islets
2.4. Secreted and Total Insulin Quantitation
2.5. RT-qPCR
2.6. Confocal Microscopy
2.7. Electron Microscopy
2.8. Immunohistochemistry
2.9. Islet Dispersion and Intracellular Calcium Imaging
2.10. Animal Care
2.11. Choice of Sexual Genders
2.12. Statistics
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thong, F.S.L.; Dugani, C.B.; Klip, A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology 2005, 4, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Zhang, L.; Brandon, A.; Wang, Q.; Begg, D.; Qi, Y.; Fu, M.; Kulkarni, R.; Teo, J.; Baldock, P.; et al. Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 2017, 6, 574–584. [Google Scholar] [CrossRef]
- Rochat, A.; Fernandez, A.; Vandromme, M.; Moles, J.P.; Bouschet, T.; Carnac, G.; Lamb, N. Insulin and Wnt1 Pathways Cooperate to Induce Reserve Cell Activation in Differentiation and Myotube Hypertrophy. Mol. Biol. Cell 2004, 15, 4544–4555. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Song, Y.L.; Xiong, C.J.; Lu, P.Q.; Xue, L.X.; Yao, C.X.; Wang, W.P.; Zhang, S.F.; Zhang, S.F.; Wei, Q.X.; et al. Insulin induces proliferation and cardiac differentiation of P19CL6 cells in a dose-dependent manner. Dev. Growth Differ. 2013, 55, 676–686. [Google Scholar] [CrossRef]
- Akoumianakis, I.; Badi, I.; Douglas, G.; Chuaiphichai, S.; Herdman, L.; Akawi, N.; Margaritis, M.; Antonopoulos, A.S.; Oikonomou, E.K.; Psarros, C.; et al. Insulin-induced vascular redox dysregulation in human atherosclerosis is ameliorated by dipeptidyl peptidase 4 inhibition. Sci. Transl. Med. 2020, 12, eaav8824. [Google Scholar] [CrossRef]
- Ito, H.; Hiroe, M.; Hirata, Y.; Tsujino, M.; Adachi, S.; Shichiri, M.; Koike, A.; Nogami, A.; Marumo, F. Insulin-like growth factor-I induces hypertrophy with increased expression of muscle specific genes in cultured rat cardiomyocytes. Circulation 1993, 87, 1715–1721. [Google Scholar] [CrossRef] [Green Version]
- Meda, P.; Schuit, F. Glucose-stimulated insulin secretion: The hierarchy of its multiple cellular and subcellular mechanisms. Diabetologia 2013, 56, 2552–2555. [Google Scholar] [CrossRef] [Green Version]
- Poitout, V. Fatty acids and insulin secretion: From FFAR and near? Diabetes 2018, 67, 1932–1934. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Brennan, L.; Bender, K. Amino acid metabolism, β-cell function, and diabetes. Diabetes 2006, 55, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef] [Green Version]
- Kang, G.; Leech, C.A.; Chepurny, O.G.; Coetzee, W.A.; Holz, G.G. Role of the cAMP sensor Epac as a determinant of KATP channel ATP sensitivity in human pancreatic β-cells and rat INS-1 cells. J. Physiol. 2008, 586, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Nakazaki, M.; Crane, A.; Hu, M.; Seghers, V.; Ullrich, S.; Aguilar-Bryan, L.; Bryan, J. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes 2002, 51, 3440–3449. [Google Scholar] [CrossRef] [Green Version]
- Nunemaker, C.S.; Bertram, R.; Sherman, A.; Tsaneva-Atanasova, K.; Daniel, C.R.; Satin, L.S. Glucose modulates [Ca2+]i oscillations in pancreatic islets via ionic and glycolytic mechanisms. Biophys. J. 2006, 91, 2082–2096. [Google Scholar] [CrossRef] [Green Version]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidenced in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [CrossRef]
- Colsoul, B.; Schraenen, A.; Lemaire, K.; Quintens, R.; Van Lommel, L.; Segal, A.; Owsianik, G.; Talavera, K.; Voets, T.; Margolskee, R.F.; et al. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc. Natl. Acad. Sci. USA 2010, 107, 5208–5213. [Google Scholar] [CrossRef] [Green Version]
- Fridlyand, L.E.; Tamarina, N.; Philipson, L.H. Bursting and calcium oscillations in pancreatic β-cells: Specific pacemakers for specific mechanisms. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E517–E532. [Google Scholar] [CrossRef] [Green Version]
- Bertram, R.; Satin, L.; Zhang, M.; Smolen, P.; Sherman, A. Calcium and glycolysis mediate multiple bursting modes in pancreatic islets. Biophys. J. 2004, 87, 3074–3087. [Google Scholar] [CrossRef] [Green Version]
- Bertram, R.; Serman, A.; Satin, L.S. Electrical Bursting, Calcium Oscillations, and Synchronization of Pancreatic Islets. Adv. Exp. Med. Biol. 2010, 654, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Song, S.H.; McIntyre, S.S.; Shah, H.; Veldhuis, J.D.; Hayes, P.C.; Butler, P.C. Direct measurement of pulsatile insulin secretion from the portal vein in human subjects. J. Clin. Endocrinol. Metab. 2000, 85, 4491–4499. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Barg, S.; Gylfe, E.; Tengholm, A. cAMP mediators of pulsatile insulin secretion from glucose-stimulated single β-cells. J. Biol. Chem. 2010, 285, 23007–23018. [Google Scholar] [CrossRef] [Green Version]
- Dyachok, O.; Idevall-Hagren, O.; Sågetorp, J.; Tian, G.; Wuttke, A.; Arrieumerlou, C.; Akusjärvi, G.; Gylfe, E.; Tengholm, A. Glucose-Induced Cyclic AMP Oscillations Regulate Pulsatile Insulin Secretion. Cell Metab. 2008, 8, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengholm, A.; Gylfe, E. Oscillatory control of insulin secretion. Mol. Cell. Endocrinol. 2009, 297, 58–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengholm, A. Cyclic AMP dynamics in the pancreatic B-cell. Upsala J. Med. Sci. 2012, 117, 355–369. [Google Scholar] [CrossRef]
- Lang, D.A.; Matthews, D.R.; Burnett, M.; Turner, M.C. Brief, Irregular Oscillations of Basal Plasma Insulin and Glucose Concentrations in Diabetic Man. Diabetes 1981, 30, 435–439. [Google Scholar] [CrossRef]
- Regard, J.B.; Kataoka, H.; Cano, D.A.; Camerer, E.; Yin, L.; Zheng, Y.; Scanlan, T.S.; Hebrok, M.; Coughlin, S.R. Probing cell type —Specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J. Clin. Investig. 2007, 117, 4034–4043. [Google Scholar] [CrossRef] [Green Version]
- Iankova, I.; Chavey, C.; Clape, C.; Colomer, C.; Gue, N.C.; Grillet, N.; Brunet, J.-F.; Annicotte, J.-S.; Fajas, L. Regulator of G Protein Signaling-4 Controls Fatty Acid and Glucose Homeostasis. Endocrinology 2008, 149, 5706–5712. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, Q.; Straub, S.G.; Sharp, G.W.G. Channel Both Gi and Go Heterotrimeric G Proteins Are Required to Exert the Full Effect of Norepinephrine on the β-Cell KATP Channel. J. Biol. Chem. 2008, 283, 5306–5316. [Google Scholar] [CrossRef] [Green Version]
- De Azua, I.R.; Scarselli, M.; Rosemond, E.; Gautam, D.; Jou, W.; Gavrilova, O.; Ebert, P.J.; Levitt, P.; Wess, J. RGS4 is a negative regulator of insulin release from pancreatic β-cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 7999–8004. [Google Scholar] [CrossRef] [Green Version]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Fagerholm, V.; Haaparanta, M.; Scheinin, M. α2-Adrenoceptor Regulation of Blood Glucose Homeostasis. Basic Clin. Pharmacol. Toxicol. 2011, 108, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wang, Y.; Park, S.; Bajpayee, N.S.; Vi, D.; Nagaoka, Y.; Birnbaumer, L.; Jiang, M. Go2 G protein mediates galanin inhibitory effects on insulin release from pancreatic β cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2636–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabets Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Strowski, M.Z.; Parmar, R.M.; Blake, A.D.; Schaeffer, J.M. Somatostatin Inhibits Insulin and Glucagon Secretion via Two Receptor Subtypes: An in Vitro Study of Pancreatic Islets from Somatostatin Receptor 2 Knockout Mice. Endocrinology 2014, 141, 111–117. [Google Scholar] [CrossRef]
- Gautam, D.; Han, S.-J.; Hamdan, F.F.; Jeon, J.; Li, B.; Li, J.H.; Cui, Y.; Mears, D.; Lu, H.; Deng, C.; et al. A critical role for β cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab. 2006, 3, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.; Butcher, A.J.; Mcwilliams, P.; Jones, D.; Wess, J.; Hamdan, F.F.; Werry, T.; Rosethorne, E.; Charlton, S.J.; Munson, S.E.; et al. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proc. Natl. Acad. Sci. USA 2010, 107, 21181–21186. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fendler, B.; Peercy, B.; Goel, P.; Bertram, R.; Sherman, A.; Satin, L.S. Long lasting synchronization of calcium oscillations by cholinergic stimulation in isolated pancreatic islets. Biophys. J. 2008, 95, 4676–4688. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, L. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 2016, 57, R93–R108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, E.; Palmer, T.M.; Yarwood, S.J. The future of EPAC-targeted therapies: Agonism versus antagonism. Trends Pharmacol. Sci. 2015, 36, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, K.J.; Coltharp, C.; Amzel, M.L.; Zhang, J. Direct Activation of Epac by Sulfonylurea is Isoform Selective. Chem. Biol. 2011, 18, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, Y.; Wang, J.; Kim, D.S.; Wu, H.; Sjögren, B.; Gao, W.; Luttrell, L.; Wang, H. Regulator of G protein signaling 2 is a key regulator of pancreatic β-cell mass and function. Cell Death Dis. 2017, 8, e2821. [Google Scholar] [CrossRef] [Green Version]
- Vivot, K.; Moullé, V.S.; Zarrouki, B.; Tremblay, C.; Mancini, A.D.; Maachi, H.; Ghislain, J.; Poitout, V. The regulator of G-protein signaling RGS16 promotes insulin secretion and β-cell proliferation in rodent and human islets. Mol. Metab. 2016, 5, 988–996. [Google Scholar] [CrossRef]
- Wang, Q.; Pronin, A.N.; Levay, K.; Almaca, J.; Fornoni, A.; Caicedo, A.; Slepak, V.Z. Regulator of G-protein signaling Gβ5-R7 is a crucial activator of muscarinic M3 receptor-stimulated insulin secretion. FASEB J. 2017, 11, 4734–4744. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Henry, T.A.N.; Pronin, A.N.; Jang, G.-F.; Lubaczeuski, C.; Crabb, J.W.; Bernal-Mizrachi, E.; Slepak, V.Z. The regulatory G protein signaling complex, Gβ5-R7, promotes glucose- and extracellular signal stimulated insulin secretion. J. Biol. Chem. 2020, 295, 7213–7223. [Google Scholar] [CrossRef] [Green Version]
- Bikopoulos, G.; Da Silva Pimenta, A.; Lee, S.C.; Lakey, J.R.; Der, S.D.; Chan, C.B.; Ceddia, R.B.; Wheeler, M.B.; Rozakis-adcock, M. Ex vivo transcriptional profiling of human pancreatic islets following chronic exposure to monounsaturated fatty acids. J. Endocrinol. 2008, 196, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Cifelli, C.; Rose, R.A.; Zhang, H.; Voigtlaender-bolz, J.; Bolz, S.; Backx, P.H.; Heximer, S.P. RGS4 Regulates Parasympathetic Signaling and Heart Rate Control in the Sinoatrial Node. Circ. Res. 2008, 103, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Bastin, G.; Singh, K.; Dissanayake, K.; Mighiu, A.S.; Nurmohamed, A.; Heximer, S.P. Amino-terminal Cysteine Residues Differentially Influence RGS4 Protein Plasma Membrane Targeting, Intracellular Trafficking, and Function. J. Biol. Chem. 2012, 287, 28966–28974. [Google Scholar] [CrossRef] [Green Version]
- Bastin, G.; Dissanayake, K.; Langburt, D.; Tam, A.L.C.; Lee, S.H.; Lachhar, K.; Heximer, S.P. RGS4 controls Gαi3-mediated regulation of Bcl-2 phosphorylation on TGN38-containing intracellular membranes. J. Cell Sci. 2020, 133, jcs241034. [Google Scholar] [CrossRef] [PubMed]
- Hardy, A.B.; Prentice, K.J.; Froese, S.; Liu, Y.; Andrews, G.K.; Wheeler, M.B. Zip4 mediated zinc influx stimulates insulin secretion in pancreatic beta cells. PLoS ONE 2015, 10, e0119136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, Z.; Yau, D.; Chan, F.; LeRoith, D.; Chan, C.B.; Wheeler, M.B. Insulin resistance causes increased beta-cell mass but defective glucose-stimulated insulin secretion in a murine model of type 2 diabetes. Diabetologia 2006, 49, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Robson-Doucette, C.A.; Wheeler, M.B. Uncoupling protein 2 regulates reactive oxygen species formation in islets and influences susceptibility to diabetogenic action of streptozotocin. J. Endocrinol. 2009, 203, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, A.B.; Fox, J.E.M.; Giglou, P.R.; Wijesekara, N.; Bhattacharjee, A.; Sultan, S.; Gyulkhandanyan, A.V.; Gaisano, H.Y.; MacDonald, P.E.; Wheeler, M.B. Characterization of Erg K+ channels in α- and β-cells of mouse and human islets. J. Biol. Chem. 2009, 284, 30441–30452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Gu, S.; Al-Sabeq, B.; Wang, S.; He, J.; Tam, A.; Cifelli, C.; Mathalone, N.; Tirgari, S.; Boyd, S.; et al. Origin-specific epigenetic program correlates with vascular bed-specific differences in Rgs5 expression. FASEB J. 2012, 26, 181–191. [Google Scholar] [CrossRef]
- Pacey, L.K.K.; Xuan, I.C.Y.; Guan, S.; Sussman, D.; Henkelman, R.M.; Chen, Y.; Thomsen, C.; Hampson, D.R. Delayed myelination in a mouse model of fragile X syndrome. Hum. Mol. Genet. 2013, 22, 3920–3930. [Google Scholar] [CrossRef]
- Hastoy, B.; Clark, A.; Rorsman, P.; Lang, J. Fusion pore in exocytosis: More than an exit gate? A β-cell perspective. Cell Calcium 2017, 68, 45–61. [Google Scholar] [CrossRef]
- Untereiner, A.; Abdo, S.; Bhattacharjee, A.; Gohil, H.; Pourasgari, F.; Ibeh, N.; Lai, M.; Batchuluun, B.; Wong, A.; Khuu, N.; et al. GABA promotes β-cell proliferation, but does not overcome impaired glucose homeostasis associated with diet-induced obesity. FASEB J. 2019, 33, 3968–3984. [Google Scholar] [CrossRef]
- Ebert, P.J.; Campbell, D.B.; Levitt, P. BAC Transgenic Analysis of Dynamic Expression Patterns of Regulator of G-Protein Signaling 4 (RGS4) During Development. I. Cerebral Cortex. Neuroscience 2007, 142, 1145–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goginashvili, A.; Zhang, Z.; Erbs, E.; Spiegelhalter, C.; Kessler, P.; Mihlan, M.; Pasquier, A.; Krupina, K.; Schieber, N.; Cinque, L.; et al. Insulin secretory granules control autophagy in Pancreatic β cells. Science 2015, 347, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Bailey, C.L.; Fueger, P.T.; Newgard, C.B.; Casey, P.J.; Kimple, M.E. Rap1 Promotes Multiple Pancreatic Islet Cell Functions and Signals through Mammalian Target of Rapamycin Complex 1 to Enhance Proliferation. J. Biol. Chem. 2010, 285, 15777–15785. [Google Scholar] [CrossRef] [Green Version]
- Tanemura, M.; Ohmura, Y.; Deguchi, T.; Machida, T.; Tsukamoto, R.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Ito, T.; et al. Rapamycin Causes Upregulation of Autophagy and Impairs Islets Function Both In Vitro and In Vivo. Am. J. Transplant. 2012, 12, 102–114. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M. V TOR-centric view on insulin resistance and diabetic complications: Perspective for endocrinologists and gerontologists. Cell Death Dis. 2013, 4, e964–e968. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Tan-sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation induced autophagy through TORC1 inhibition. Mol. Cell 2015, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Zhao, A.; Ohara-imaizumi, M.; Brissova, M.; Benninger, R.K.P.; Xu, Y.; Hao, Y.; Abramowitz, J.; Boulay, G.; Powers, A.C.; Piston, D.; et al. Gαo Represses Insulin Secretion by Reducing Vesicular Docking in Pancreatic β-Cells. Diabetes 2010, 59, 2522–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katada, T.; Ui, M. Perfusion of the Pancreas Isolated from Petrussis-Sensitized Rats: Potentiation of Insulin Secretory Responses Due to β-Adrenergic Stimulation. Endocrinology 1977, 101, 1247–1255. [Google Scholar] [CrossRef]
- Marty, C.; Ye, R.D. Heterotrimeric G Protein Signaling Outside the Realm of Seven Transmembrane Domain Receptors. Mol. Pharmacol. 2010, 78, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Hepler, J.R.; Gilman, A.G.; Mumby, S.M. Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc. Natl. Acad. Sci. USA 1997, 94, 6159–6163. [Google Scholar] [CrossRef] [Green Version]
- Landa, L.E.; Harbeck, M.; Kaihara, K.; Chepurny, O.; Kitiphongspattana, K.; Graf, O.; Nikolaev, V.O.; Lohse, M.J.; Holz, G.G.; Roe, M.W. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 β-cell line. J. Biol. Chem. 2005, 280, 31294–31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, E.L.; Schwartzman, R.A.; Bond, M.; Insel, P.A. Multi-tasking RGS Proteins in the Heart: The Next Therapeutic Target ? Circ. Res. 2005, 96, 401–411. [Google Scholar] [CrossRef]
- Xie, G.; Pamler, P.P. How regulators of G protein signaling achieve selective regulation. J. Mol. Biol 2007, 366, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Popov, S.G.; Krishna, U.M.; Falck, J.R.; Wilkie, T.M. Ca2+/Calmodulin Reverses Phosphatidylinositol 3, 4, 5-Trisphosphate-dependent Inhibition of Regulators of G Protein-signaling GTPase-activating Protein Activity. J. Biol. Chem. 2000, 275, 18962–18968. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Inanobe, A.; Kurachi, Y. PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G protein cycle in a cardiac K+ channel. Proc. Natl. Acad. Sci. USA 2002, 99, 4325–4330. [Google Scholar] [CrossRef] [Green Version]
- Bouche, C.; Lopez, X.; Fleischman, A.; Cypess, A.M.; Shea, S.O.; Stefanovski, D.; Bergman, R.N.; Rogatsky, E.; Stein, D.T.; Kahn, R.C.; et al. Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proc. Natl. Acad. Sci. USA 2010, 107, 4770–4775. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Bruning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic β Cells Creates an Insulin Secretory Defect Similar to that in Type 2 Diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Popov, S.; Bera, A.K.; Wilkie, T.M.; Muallem, S. RGS proteins provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol. Cell 2001, 7, 651–660. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastin, G.; Luu, L.; Batchuluun, B.; Mighiu, A.; Beadman, S.; Zhang, H.; He, C.; Al Rijjal, D.; Wheeler, M.B.; Heximer, S.P. RGS4-Deficiency Alters Intracellular Calcium and PKA-Mediated Control of Insulin Secretion in Glucose-Stimulated Beta Islets. Biomedicines 2021, 9, 1008. https://doi.org/10.3390/biomedicines9081008
Bastin G, Luu L, Batchuluun B, Mighiu A, Beadman S, Zhang H, He C, Al Rijjal D, Wheeler MB, Heximer SP. RGS4-Deficiency Alters Intracellular Calcium and PKA-Mediated Control of Insulin Secretion in Glucose-Stimulated Beta Islets. Biomedicines. 2021; 9(8):1008. https://doi.org/10.3390/biomedicines9081008
Chicago/Turabian StyleBastin, Guillaume, Lemieux Luu, Battsetseg Batchuluun, Alexandra Mighiu, Stephanie Beadman, Hangjung Zhang, Changhao He, Dana Al Rijjal, Michael B. Wheeler, and Scott P. Heximer. 2021. "RGS4-Deficiency Alters Intracellular Calcium and PKA-Mediated Control of Insulin Secretion in Glucose-Stimulated Beta Islets" Biomedicines 9, no. 8: 1008. https://doi.org/10.3390/biomedicines9081008
APA StyleBastin, G., Luu, L., Batchuluun, B., Mighiu, A., Beadman, S., Zhang, H., He, C., Al Rijjal, D., Wheeler, M. B., & Heximer, S. P. (2021). RGS4-Deficiency Alters Intracellular Calcium and PKA-Mediated Control of Insulin Secretion in Glucose-Stimulated Beta Islets. Biomedicines, 9(8), 1008. https://doi.org/10.3390/biomedicines9081008