
CD40 Agonist Monoclonal Antibody-Mediated Hepatitis in TNF-Receptor 1 Gene Knockout Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. TNFR1−/− Mice and Induction of CD40 mAb-Mediated Hepatitis
2.2. Experimental Protocol and Tissue Sampling
2.3. Real Time PCR
2.4. Microarray
2.5. Serum Analyses
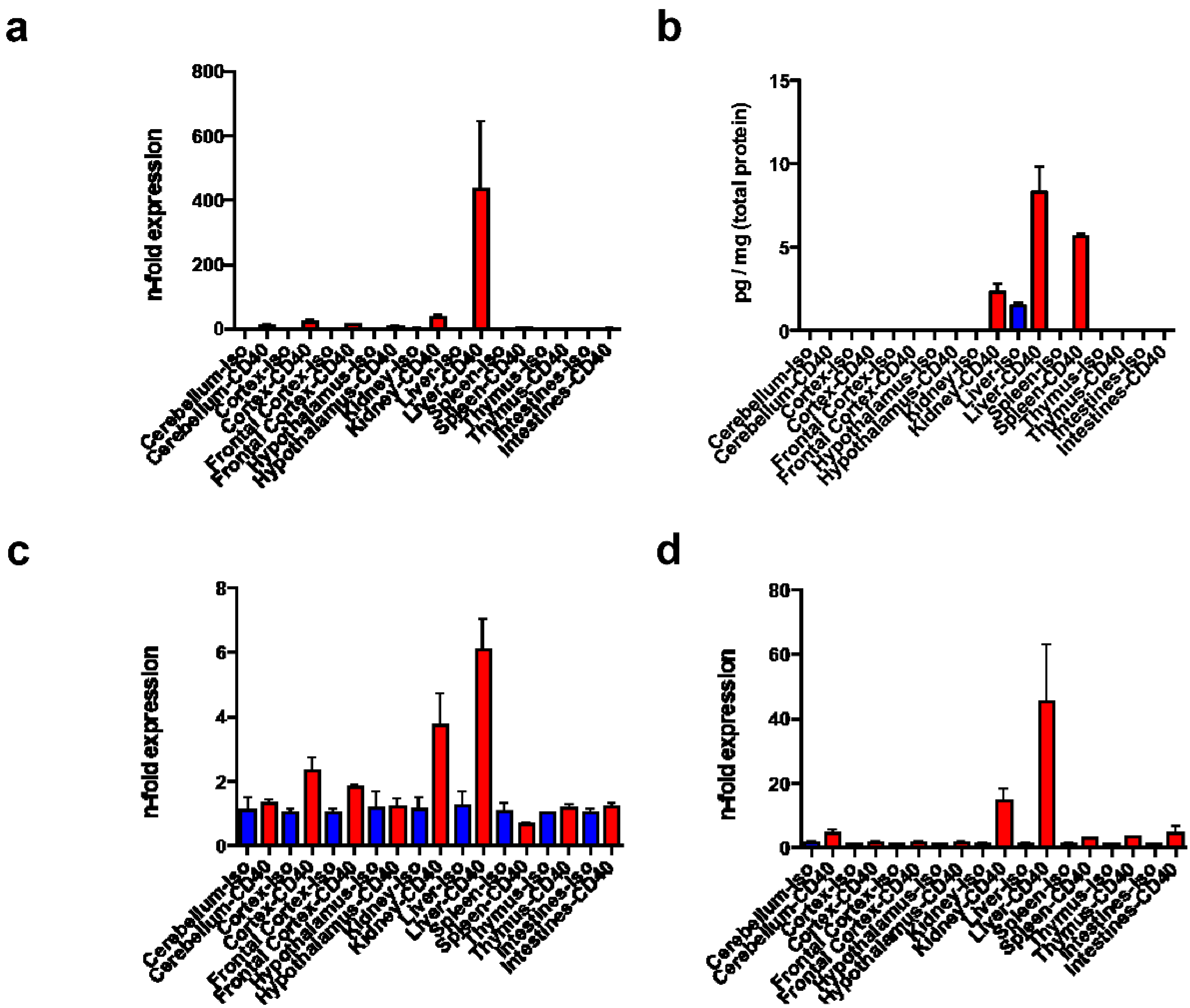
2.6. Liver Tissue TNF-α ELISA
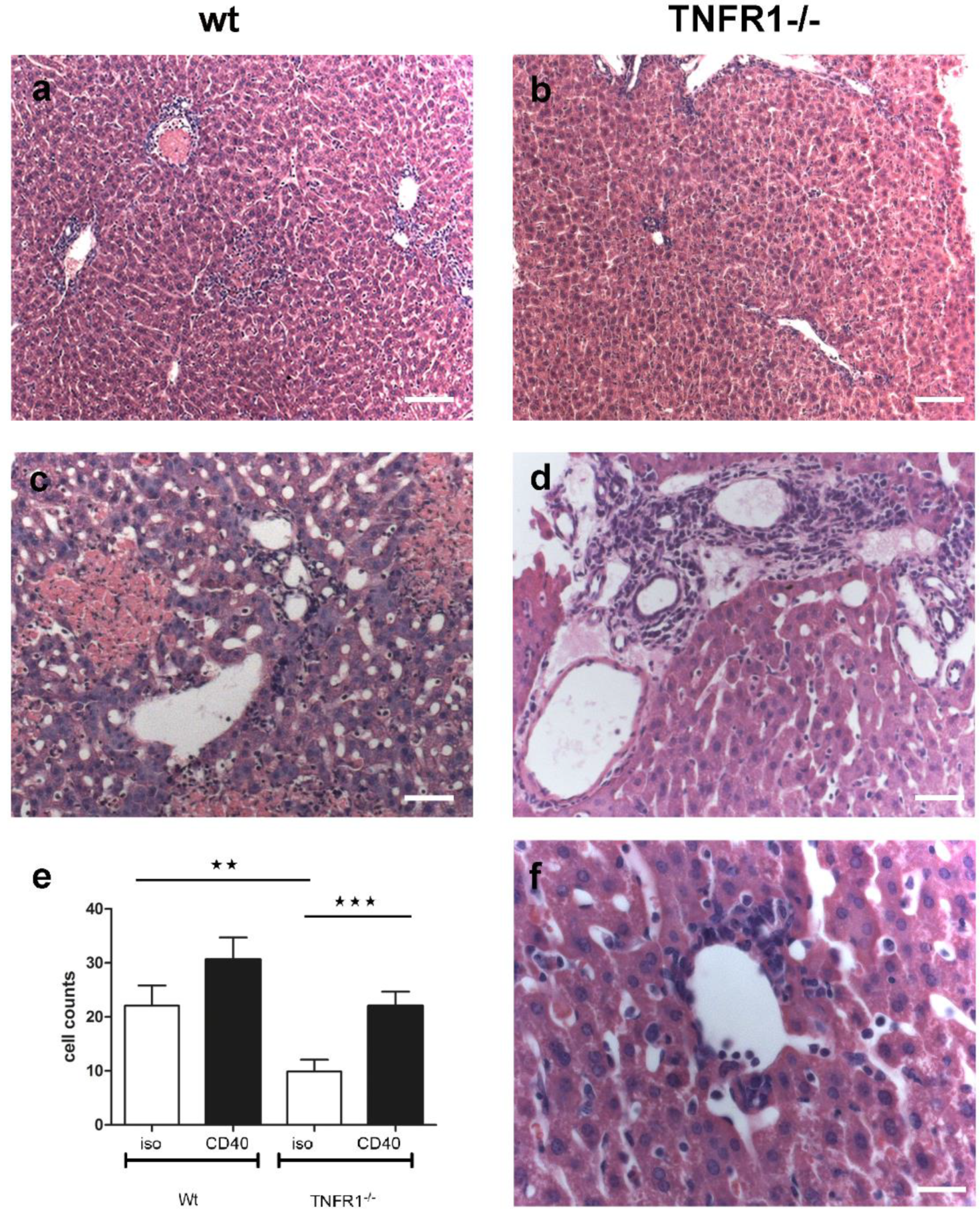
2.7. Histological and Quantitative Analysis of Liver Tissue
2.8. Statistical Analysis
3. Results
3.1. CD40 mAB Induced Sickness Effects in wt and TNFR1−/− Mice
3.2. Tissue Distribution of tnf, tnfr1, and cd40
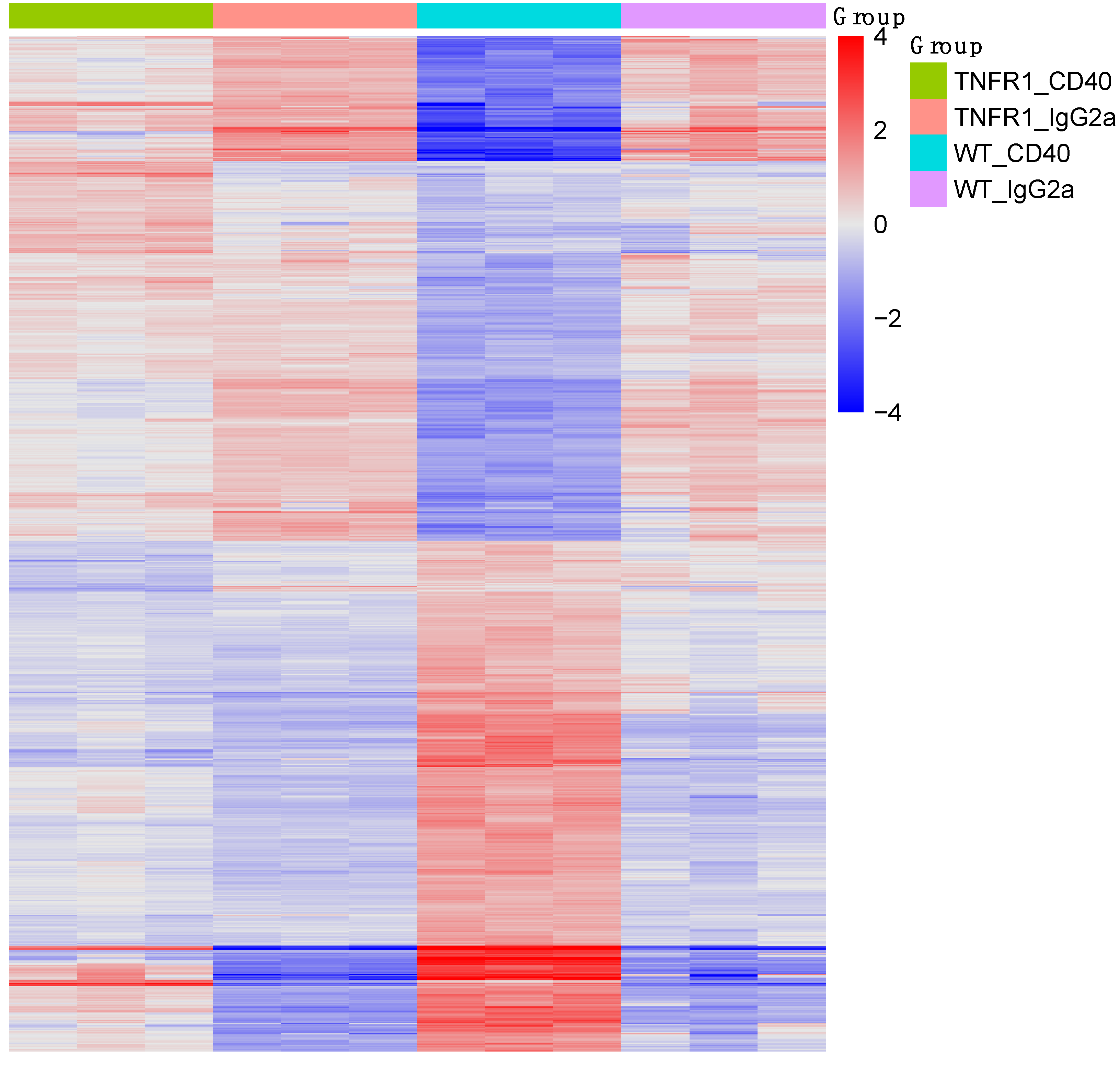
3.3. Gene Expression Microarray Analysis of Liver Tissue
3.4. Serum Levels of sALT, BUN and GLU
3.5. Histological Analysis of Hepatic Inflammation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munroe, M.E. Functional roles for T cell CD40 in infection and autoimmune disease: The role of CD40 in lymphocyte homeostasis. Semin. Immunol. 2009, 21, 283–288. [Google Scholar] [CrossRef]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [Green Version]
- Djureinovic, D.; Wang, M.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef] [PubMed]
- Pryce, C.R.; Fontana, A. Depression in autoimmune diseases. Curr. Top. Behav. Neurosci. 2017, 31, 139–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vial, G.; Gensous, N.; Duffau, P. The CD40-CD40L axis: Current and future implications in clinical immunology. Rev. Med. Interne 2021, 2. [Google Scholar] [CrossRef]
- Zhou, F.; Ajuebor, M.N.; Beck, P.L.; Le, T.; Hogaboam, C.M.; Swain, M.G. CD154-CD40 interactions drive hepatocyte apoptosis in murine fulminant hepatitis. Hepatology 2005, 42, 372–380. [Google Scholar] [CrossRef]
- Kimura, K.; Moriwaki, H.; Nagaki, M.; Saio, M.; Nakamoto, Y.; Naito, M.; Kuwata, K.; Chisari, F.V. Pathogenic role of B cells in anti-CD40-induced necroinflammatory liver disease. Am. J. Pathol. 2006, 168, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Chatzigeorgiou, A.; Lyberi, M.; Chatzilymperis, G.; Nezos, A.; Kamper, E. CD40/CD40L signaling and its implication in health and disease. Biofactors 2009, 35, 474–483. [Google Scholar] [CrossRef]
- Advani, R.; Forero-Torres, A.; Furman, R.R.; Rosenblatt, J.D.; Younes, A.; Ren, H.; Harrop, K.; Whiting, N.; Drachman, J.G. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J. Clin. Oncol. 2009, 27, 4371–4377. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Forero-Torres, A.; Shustov, A.; Drachman, J.G. A phase I study of dacetuzumab (SGN-40, a humanized anti-CD40 monoclonal antibody) in patients with chronic lymphocytic leukemia. Leuk Lymphoma 2010, 51, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Bartlett, N.; Beaven, A.; Myint, H.; Nasta, S.; Northfelt, D.W.; Whiting, N.C.; Drachman, J.G.; Lobuglio, A.F.; Moskowitz, C.H. Pilot study of dacetuzumab in combination with rituximab and gemcitabine for relapsed or refractory diffuse large B-cell lymphoma. Leuk Lymphoma 2013, 54, 277–283. [Google Scholar] [CrossRef] [PubMed]
- De Vos, S.; Forero-Torres, A.; Ansell, S.M.; Kahl, B.; Cheson, B.D.; Bartlett, N.L.; Furman, R.R.; Winter, J.N.; Kaplan, H.; Timmerman, J.; et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J. Hematol. Oncol. 2014, 44. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef] [Green Version]
- Machiels, J.P.; Gomez-Roca, C.; Michot, J.M.; Zamarin, D.; Mitchell, T.; Catala, G.; Eberst, L.; Jacob, W.; Jegg, A.M.; Cannarile, M.A.; et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother Cancer 2020, 8, e001153. [Google Scholar] [CrossRef]
- Lee, B.N.; Dantzer, R.; Langley, K.E.; Bennett, G.J.; Dougherty, P.M.; Dunn, A.J.; Meyers, C.A.; Miller, A.H.; Payne, R.; Reuben, J.M.; et al. A cytokine-based neuroimmunologic mechanism of cancer-related symptoms. Neuroimmunomodulation 2004, 11, 279–292. [Google Scholar] [CrossRef]
- Pollak, Y.; Ovadia, H.; Orion, E.; Weidenfeld, J.; Yirmiya, R. The EAE-associated behavioral syndrome: I. Temporal correlation with inflammatory mediators. J. Neuroimmunol. 2003, 137, 94–99. [Google Scholar] [CrossRef]
- Taraborrelli, C.; Palchykova, S.; Tobler, I.; Gast, H.; Birchler, T.; Fontana, A. TNFR1 is essential for CD40, but not for lipopolysaccharide-induced sickness behavior and clock gene dysregulation. Brain Behav. Immun. 2011, 25, 434–442. [Google Scholar] [CrossRef]
- Gast, H.; Müller, A.; Lopez, M.; Meier, D.; Huber, R.; Dechent, F.; Prinz, M.; Emmenegger, Y.; Franken, P.; Birchler, T.; et al. CD40 activation induces NREM sleep and modulates genes associated with sleep homeost asis. Brain Behav. Immun. 2013, 27, 133–144. [Google Scholar] [CrossRef]
- Cathomas, F.; Fuertig, R.; Sigrist, H.; Newman, G.N.; Hoop, V.; Bizzozzero, M.; Mueller, A.; Luippold, A.; Ceci, A.; Hengerer, B.; et al. Pryce CR8 CD40-TNF activation in mice induces extended sickness behavior syndrome co-incident with but not dependent on activation of the kynurenine pathway. Brain Behav. Immun. 2015, 50, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Oo, Y.H.; Shetty, S.; Adams, D.H. The role of chemokines in the recruitment of lymphocytes to the liver. Dig. Dis. 2010, 28, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Medina-Echeverz, J.; Ma, C.; Duffy, A.G.; Eggert, T.; Hawk, N.; Kleiner, D.E.; Korangy, F.; Greten, T.F. Systemic agonistic anti-cd40 treatment of tumor-bearing mice modulates hepatic myeloid-suppressive cells and causes immune-mediated liver damage. Cancer Immunol. Res. 2015, 3, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingoglia, G.; Yalamanoglu, A.; Pfefferlé, M.; Dubach, I.L.; Schaer, C.A.; Valkova, K.; Hansen, K.; Schulthess, N.; Humar, R.; Schaer, D.J.; et al. Line-selective macrophage activation with an anti-CD40 antibody drives a hemophagocytic syndrome in mice. Blood Adv. 2020, 4, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.F.; Strauss, L.; Greter, M.; Gast, H.; Recher, M.; Becher, B.; Fontana, A. Neutralization of colony-stimulating factor 1 receptor prevents sickness behavior syndrome by reprogramming inflammatory monocytes to produce IL-10. Brain Behav. Immun. 2015, 48, 78–85. [Google Scholar] [CrossRef]
- Rolink, A.; Melchers, F.; Andersson, J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity 1996, 5, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Petrzilka, S.; Taraborrelli, C.; Cavadini, G.; Fontana, A.; Birchler, T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J. Biol. Rhythms 2009, 24, 283–294. [Google Scholar] [CrossRef]
- Cavadini, G.; Petrzilka, S.; Kohler, P.; Jud, C.; Tobler, I.; Birchler, T.; Fontana, A. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12843–12848. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.bioconductor.org (accessed on 7 May 2021).
- Guangchuang, Y.; Wang, L.G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar]
- Zhang, S.; Breidenbach, J.D.; Russell, B.H.; George, J.; Haller, S.T. CD40/CD40L Signaling as a Promising Therapeutic Target for the Treatment of Renal Disease. J. Clin. Med. 2020, 9, 3653. [Google Scholar] [CrossRef]
- Baratta, J.L.; Ngo, A.; Lopez, B.; Kasabwalla, N.; Longmuir, K.J.; Robertson, R.T. Cellular organization of normal mouse liver: A histological, quantitative immunocytochemical, and fine structural analysis. Histochem. Cell Biol. 2009, 131, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fütterer, A.; Mink, K.; Luz, A.; Kosco-Vilbois, M.H.; Pfeffer, K. Lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 1998, 9, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Yang, G.X.; Iwakoshi, N.; Knechtle, S.J.; Kawata, K.; Tsuneyama, K.; Leung, P.; Coppel, R.L.; Ansari, A.A.; Joh, T.; et al. Anti-CD40 ligand monoclonal antibody delays the progression of murine autoimmune cholangitis. Clin. Exp. Immunol. 2013, 174, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Klaus, F.; Paterna, J.-C.; Marzorati, E.; Sigrist, H.; Götze, L.; Schwendener, S.; Bergamini, G.; Jehli, E.; Azzinnari, D.; Fuertig, R.; et al. Differential effects of peripheral and brain tumor necrosis factor on inflammation, sickness, emotional behavior and memory in mice. Brain Behav. Immun 2016, 58, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Aarts, S.; Reiche, M.; den Toom, M.; Gijbels, M.; Beckers, L.; Gerdes, N.; Lutgens, E. Depletion of CD40 on CD11c+ cells worsens the metabolic syndrome and ameliorates hepatic inflammation during NASH. Sci. Rep. 2019, 9, 14702. [Google Scholar] [CrossRef] [Green Version]
- Bojadzic, D.; Chen, J.; Alcazar, O.; Buchwald, P. Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction. Molecules 2018, 23, 1153. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Over-Expressed | FDR | Under-Expressed Genes | FDR |
|---|---|---|---|
| Ascorbate and aldarate metabolism | 3.52 × 10−9 | Protein processing in endoplasmic reticulum | 3.52 × 10−9 |
| Steroid hormone biosynthesis | 3.52 × 10−9 | Ribosome | 2.34 × 10−7 |
| Retinol metabolism | 3.52 × 10−9 | Cytokine-cytokine receptor interaction | 3.38 × 10−6 |
| Metabolism of xenobiotics by cytochrome P450 | 3.52 × 10−9 | NF-kappa B signaling pathway | 3.98 × 10−6 |
| Drug metabolism—cytochrome P450 | 3.52 × 10−9 | IL-17 signaling pathway | 6.53 × 10−6 |
| Drug metabolism—other enzymes | 3.52 × 10−9 | TNF signaling pathway | 1.67 × 10−5 |
| Bile secretion | 3.52 × 10−9 | NOD-like receptor signaling pathway | 4.22 × 10−5 |
| Chemical carcinogenesis | 3.52 × 10−9 | N-Glycan biosynthesis | 2.05 × 10−4 |
| Linoleic acid metabolism | 1.47 × 10−8 | Protein export | 2.17 × 10−4 |
| Olfactory transduction | 1.89 × 10−8 | Viral protein interaction with cytokines and cytokine rec. | 2.96 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raabe, O.; Birchler, T.; Rehrauer, H.; Eppler, E. CD40 Agonist Monoclonal Antibody-Mediated Hepatitis in TNF-Receptor 1 Gene Knockout Mice. Biomedicines 2021, 9, 863. https://doi.org/10.3390/biomedicines9080863
Raabe O, Birchler T, Rehrauer H, Eppler E. CD40 Agonist Monoclonal Antibody-Mediated Hepatitis in TNF-Receptor 1 Gene Knockout Mice. Biomedicines. 2021; 9(8):863. https://doi.org/10.3390/biomedicines9080863
Chicago/Turabian StyleRaabe, Oksana, Thomas Birchler, Hubert Rehrauer, and Elisabeth Eppler. 2021. "CD40 Agonist Monoclonal Antibody-Mediated Hepatitis in TNF-Receptor 1 Gene Knockout Mice" Biomedicines 9, no. 8: 863. https://doi.org/10.3390/biomedicines9080863
APA StyleRaabe, O., Birchler, T., Rehrauer, H., & Eppler, E. (2021). CD40 Agonist Monoclonal Antibody-Mediated Hepatitis in TNF-Receptor 1 Gene Knockout Mice. Biomedicines, 9(8), 863. https://doi.org/10.3390/biomedicines9080863