
Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape

 , and
, and 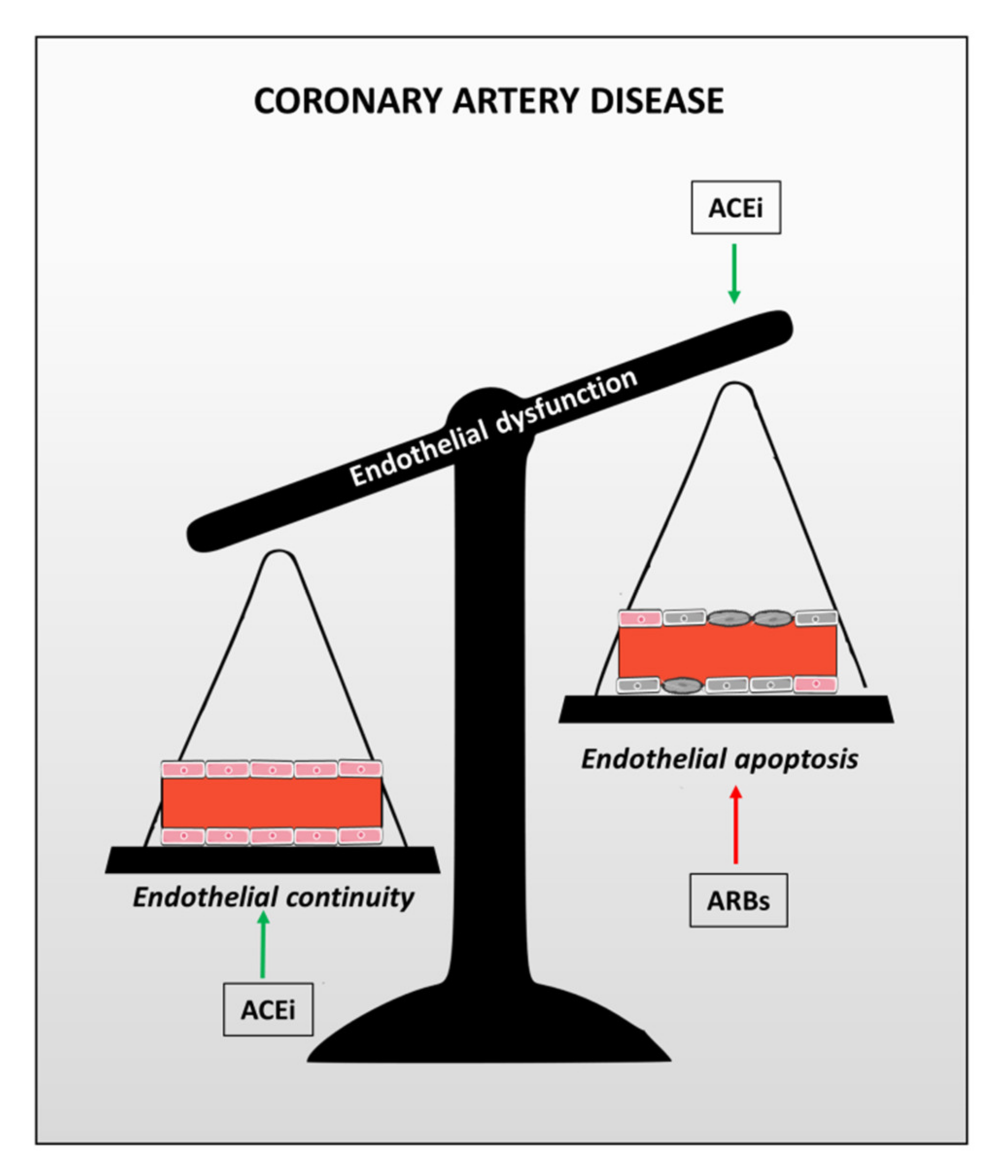{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Premise
2. Endothelium as a Barometer of Cardiovascular Disease
2.1. The Importance of Notch Signaling in the Endothelium
2.2. The Importance of the Endothelium Continuity
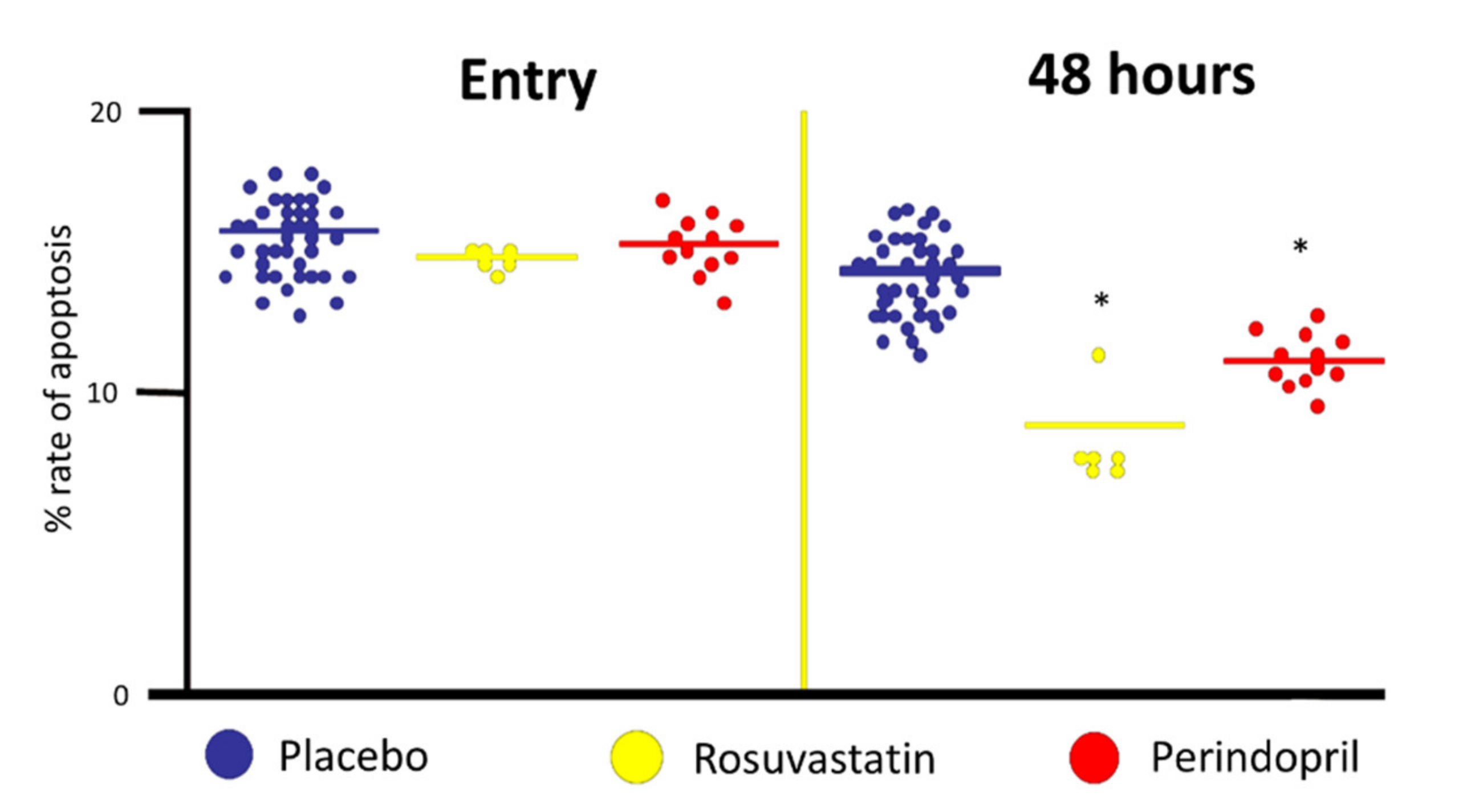
3. Focus on Ace Inhibitors (ACEi)
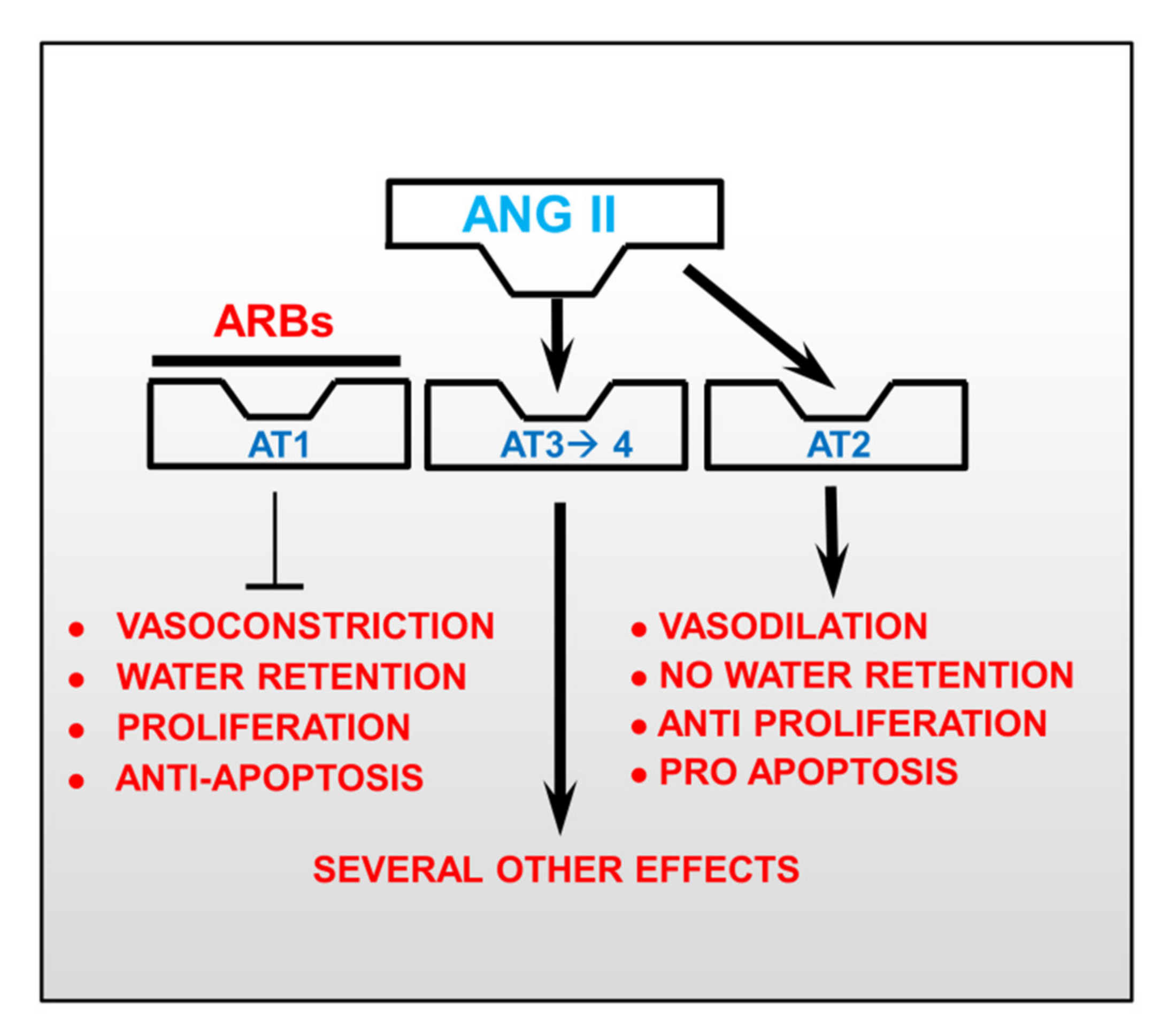
4. Focus on Angiotensin II Receptor Blockers (ARBs)
5. Focus on Lipid Lowering Agents
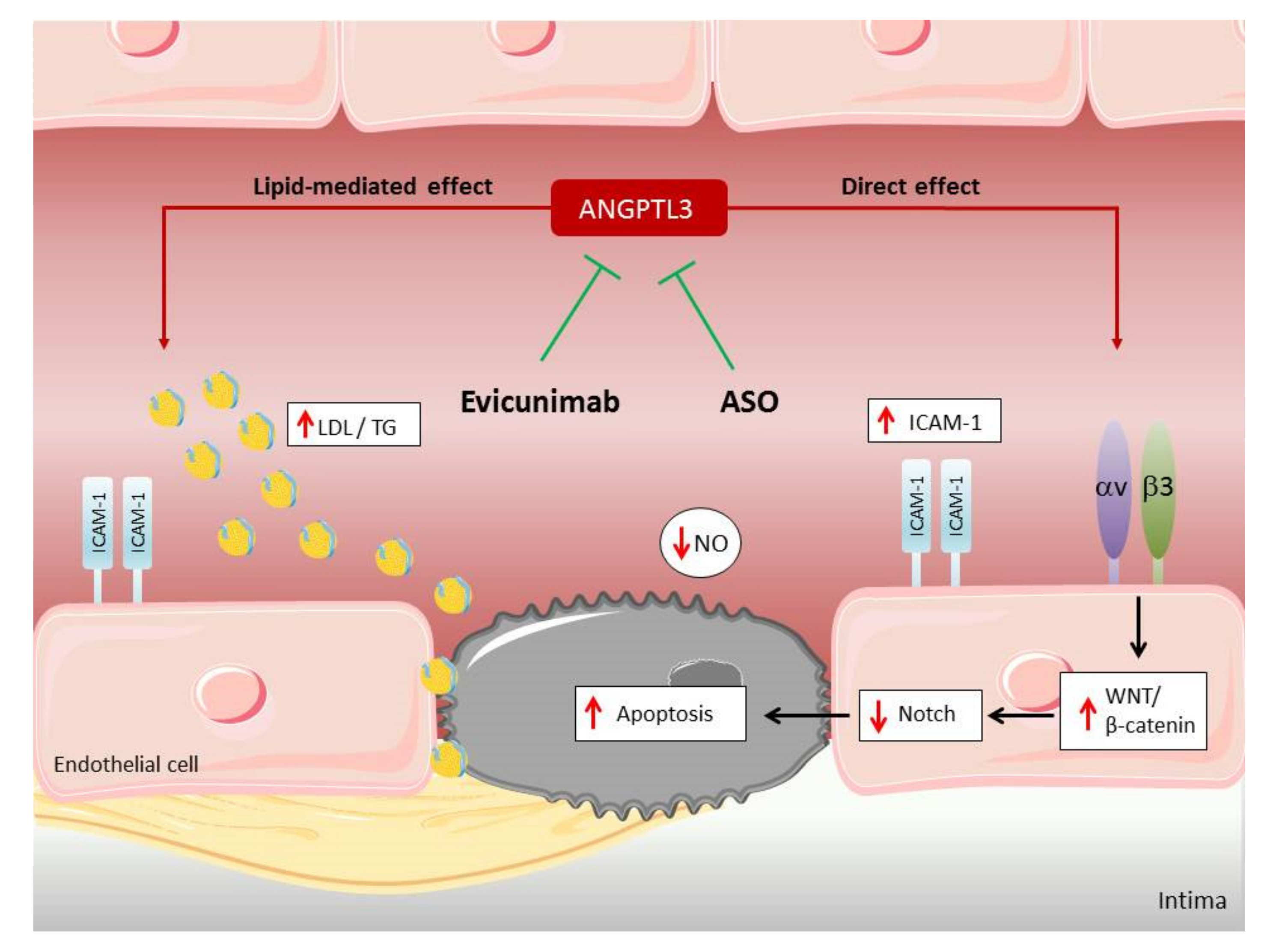
6. Focus on Angiopoietin-like Proteins (ANGPTL3) Inhibitors
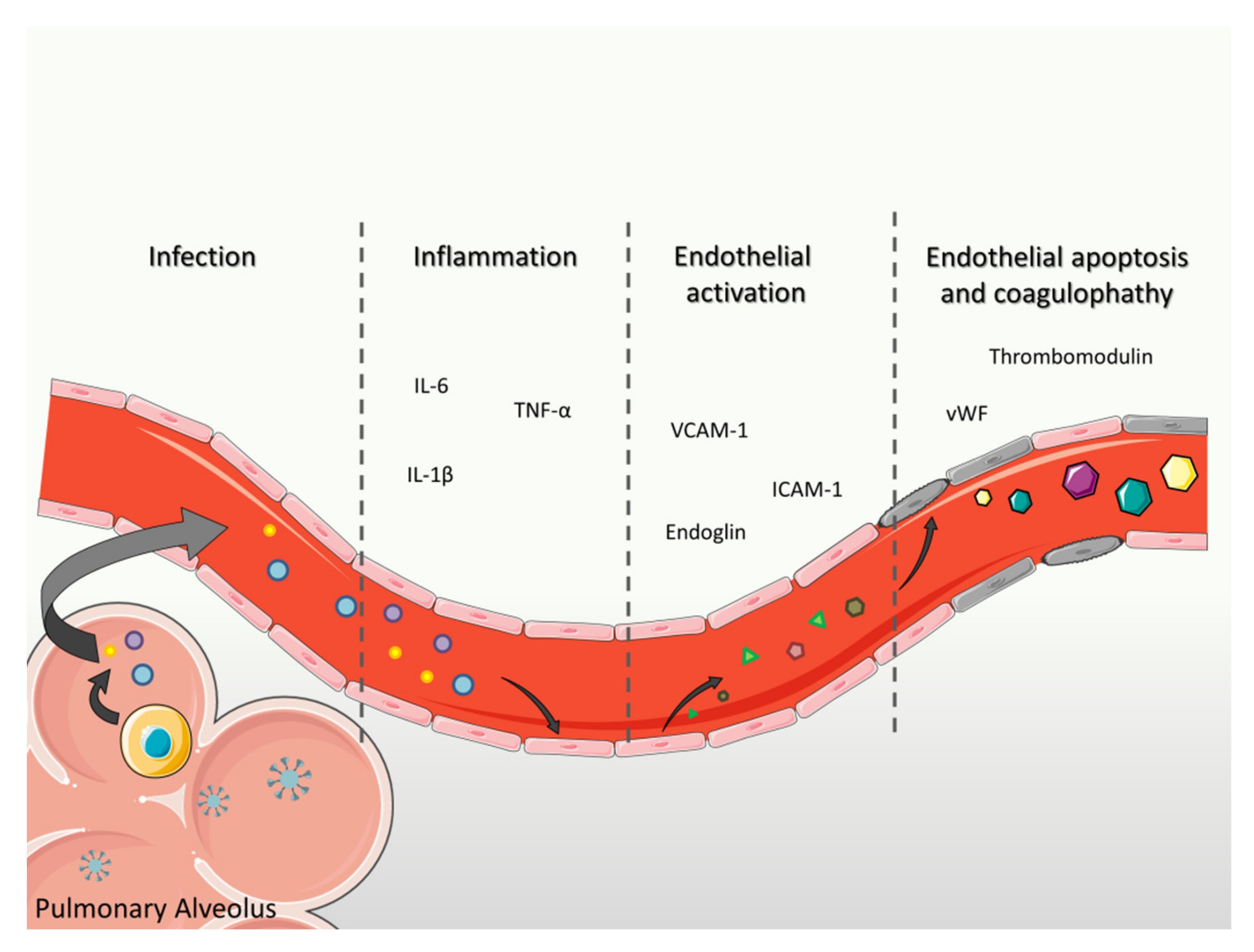
7. Importance of Endothelial Dysfunction in the COVID-19 Pneumonia
7.1. Focus on Direct Endothelium Infection by SARS-CoV-2
7.2. Focus on the Endothelium Inflammation Caused by SARS-CoV-2
7.3. Focus on Biomarkers of Endothelial Function in COVID-19
7.4. Focus on Endothelial Apoptosis in COVID-19 Patients
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferrari, R.; Guardigli, G.; Ceconi, C. Secondary prevention of CAD with ACE inhibitors: A struggle between life and death of the endothelium. Cardiovasc. Drugs Ther. 2010, 24, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Grumbach, I.M.; Fukai, T.; Cutchins, A.; Harrison, D.G. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J. Biol. Chem. 2004, 279, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Walpola, P.L.; Gotlieb, A.I.; Cybulsky, M.I.; Langille, B.L. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2–10. [Google Scholar] [CrossRef]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Mollace, V.; Gliozzi, M.; Musolino, V.; Carresi, C.; Muscoli, S.; Mollace, R.; Tavernese, A.; Gratteri, S.; Palma, E.; Morabito, C.; et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int. J. Cardiol. 2015, 184, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Aquila, G.; Fortini, F.; Vaccarezza, M.; Secchiero, P.; Rizzo, P.; Campo, G. Context-dependent function of ROS in the vascular endothelium: The role of the Notch pathway and shear stress. Biofactors 2017, 43, 475–485. [Google Scholar] [CrossRef]
- Aquila, G.; Morelli, M.B.; Vieceli Dalla Sega, F.; Fortini, F.; Nigro, P.; Caliceti, C.; Ferracin, M.; Negrini, M.; Pannuti, A.; Bonora, M.; et al. Heart rate reduction with ivabradine in the early phase of atherosclerosis is protective in the endothelium of ApoE-deficient mice. J. Physiol. Pharmacol. 2018, 69, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Fortini, F.; Aquila, G.; Campo, G.; Vaccarezza, M.; Rizzo, P. Notch signaling regulates immune responses in atherosclerosis. Front. Immunol. 2019, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Nigro, P.; Rizzo, P.; Ferrari, R. ROS, Notch, and Wnt signaling pathways: Crosstalk between three major regulators of cardiovascular biology. BioMed Res. Int. 2014, 2014, 318714. [Google Scholar] [CrossRef] [Green Version]
- Chapouly, C.; Hollier, P.L.; Guimbal, S.; Cornuault, L.; Gadeau, A.P.; Renault, M.A. Desert hedgehog-driven endothelium integrity is enhanced by gas1 (growth arrest-specific 1) but negatively regulated by Cdon (cell adhesion molecule-related/downregulated by oncogenes). Arterioscler. Thromb. Vasc. Biol. 2020, 40, e336–e349. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, G.T.K.; Hong, W. Role of hippo pathway-YAP/TAZ signaling in angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Chang, H.J.; Sung, J.M.; Park, H.B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of Statins on Coronary Atherosclerotic Plaques: The PARADIGM Study. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Schölkens, B.A.; Landgraf, W. ACE inhibition and atherogenesis. Can. J. Physiol. Pharmacol. 2002, 80, 354–359. [Google Scholar] [CrossRef]
- Rizzo, P.; Miele, L.; Ferrari, R. The Notch pathway: A crossroad between the life and death of the endothelium. Eur. Heart J. 2013, 34, 2504–2509. [Google Scholar] [CrossRef] [Green Version]
- Aquila, G.; Kostina, A.; Vieceli Dalla Sega, F.; Shlyakhto, E.; Kostareva, A.; Marracino, L.; Ferrari, R.; Rizzo, P.; Malaschicheva, A. The Notch pathway: A novel therapeutic target for cardiovascular diseases? Expert Opin. Ther. Targets 2019, 23, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef] [PubMed]
- Theodoris, C.V.; Li, M.; White, M.P.; Liu, L.; He, D.; Pollard, K.S.; Bruneau, B.G.; Srivastava, D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 2015, 160, 1072–1086. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Devallière, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef] [Green Version]
- Fortini, F.; Vieceli Dalla Sega, F.; Caliceti, C.; Aquila, G.; Pannella, M.; Pannuti, A.; Miele, L.; Ferrari, R.; Rizzo, P. Estrogen receptor β-dependent Notch1 activation protects vascular endothelium against tumor necrosis factor α (TNFα)-induced apoptosis. J. Biol. Chem. 2017, 292, 18178–18191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, F.; Vieceli Dalla Sega, F.; Caliceti, C.; Lambertini, E.; Pannuti, A.; Peiffer, D.S.; Balla, C.; Rizzo, P. Estrogen-mediated protection against coronary heart disease: The role of the Notch pathway. J. Steroid Biochem. Mol. Biol. 2019, 189, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Coupel, S.; Coulon, F.; Fitau, J.; Chatelais, M.; Cuturi, M.C.; Chiffoleau, E.; Charreau, B. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: Implication for transplant arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2258–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. [Google Scholar] [CrossRef] [Green Version]
- Briot, A.; Civelek, M.; Seki, A.; Hoi, K.; Mack, J.J.; Lee, S.D.; Kim, J.; Hong, C.; Yu, J.; Fishbein, G.A.; et al. Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J. Exp. Med. 2015, 212, 2147–2163. [Google Scholar] [CrossRef] [Green Version]
- Caliceti, C.; Aquila, G.; Pannella, M.; Morelli, M.B.; Fortini, C.; Pinton, P.; Bonora, M.; Hrelia, S.; Pannuti, A.; Miele, L.; et al. 17β-estradiol enhances signalling mediated by VEGF-A-delta-like ligand 4-notch1 axis in human endothelial cells. PLoS ONE 2013, 8, e71440. [Google Scholar] [CrossRef] [Green Version]
- Norum, H.M.; Gullestad, L.; Abraityte, A.; Broch, K.; Aakhus, S.; Aukrust, P.; Ueland, T. Increased serum levels of the notch ligand DLL1 are associated with diastolic dysfunction, reduced exercise capacity, and adverse outcome in chronic heart failure. J. Card. Fail. 2016, 22, 218–223. [Google Scholar] [CrossRef]
- Rizzo, P.; Vieceli Dalla Sega, F.; Fortini, F.; Marracino, L.; Rapezzi, C.; Ferrari, R. COVID-19 in the heart and the lungs: Could we “Notch” the inflammatory storm? Basic Res. Cardiol. 2020, 115, 31. [Google Scholar] [CrossRef] [PubMed]
- Breikaa, R.M.; Lilly, B. The Notch Pathway: A Link between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. Med. 2021, 8, 681948. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Iaconelli, M.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Bonadonna, L.; Lucentini, L.; Suffredini, E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Sci. Total Environ. 2020, 736, 139652. [Google Scholar] [CrossRef]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Delli Ponti, R.; Tartaglia, G.G. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- Harb, H.; Benamar, M.; Lai, P.S.; Contini, P.; Griffith, J.W.; Crestani, E.; Schmitz-Abe, K.; Chen, Q.; Fong, J.; Marri, L.; et al. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity 2021, 54, 1186–1199.e7. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, E.; Marchesini, J.; Campo, G.; Francolini, G.; Fortini, C.; Carrà, G.; Miccoli, M.; Ceconi, C.; Tavazzi, L.; Ferrari, R. ACE inhibition modulates endothelial apoptosis and renewal via endothelial progenitor cells in patients with acute coronary syndromes. Am. J. Cardiovasc. Drugs 2011, 11, 189–198. [Google Scholar] [CrossRef]
- Ferrari, R. Coronary artery disease: Use of ACE inhibitors in stable CAD—what is the truth? Nat. Rev. Cardiol. 2014, 11, 315–316. [Google Scholar] [CrossRef]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: Role of tumor necrosis factor-alpha. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Valgimigli, M.; Agnoletti, L.; Curello, S.; Comini, L.; Francolini, G.; Mastrorilli, F.; Merli, E.; Pirani, R.; Guardigli, G.; Grigolato, P.G.; et al. Serum from patients with acute coronary syndromes displays a proapoptotic effect on human endothelial cells: A possible link to pan-coronary syndromes. Circulation 2003, 107, 264–270. [Google Scholar] [CrossRef]
- Bachetti, T.; Comini, L.; Agnoletti, L.; Pedersini, P.; Gaia, G.; Cargnoni, A.; Bellet, M.; Curello, S.; Ferrari, R. Effects of chronic noradrenaline on the nitric oxide pathway in human endothelial cells. Basic Res. Cardiol. 1998, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, C.; Francolini, G.; Bastianon, D.; Gitti, G.L.; Comini, L.; Ferrari, R. Differences in the effect of angiotensin-converting enzyme inhibitors on the rate of endothelial cell apoptosis: In vitro and in vivo studies. Cardiovasc. Drugs Ther. 2007, 21, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.H.; Hall, A.S. Angiotensin receptor blockers may increase risk of myocardial infarction: Unraveling the ARB-MI paradox. Circulation 2006, 114, 838–854. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Boersma, E. The impact of ACE inhibition on all-cause and cardiovascular mortality in contemporary hypertension trials: A review. Expert Rev. Cardiovasc. Ther. 2013, 11, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Fakheri, R.; Toklu, B.; Ogedegbe, G.; Weintraub, H.; Messerli, F.H. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers in patients without heart failure? Insights from 254,301 patients from randomized trials. Mayo Clin. Proc. 2016, 91, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Costanzo, P.; Cleland, J.G.; Vassallo, E.; Ruggiero, D.; Rosano, G.; Perrone-Filardi, P. A meta-analysis reporting effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in patients without heart failure. J. Am. Coll. Cardiol. 2013, 61, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Zhang, W.; Zhang, X.; Han, F.; Li, X.; He, X.; Li, Q.; Chen, J. Effect of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on all-cause mortality, cardiovascular deaths, and cardiovascular events in patients with diabetes mellitus: A meta-analysis. JAMA Intern. Med. 2014, 174, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Thomopoulos, C.; Parati, G.; Zanchetti, A. Effects of blood pressure lowering on outcome incidence in hypertension: 4. Effects of various classes of antihypertensive drugs--overview and meta-analyses. J. Hypertens. 2015, 33, 195–211. [Google Scholar] [CrossRef]
- Messerli, F.H.; Bangalore, S. Angiotensin receptor blockers reduce cardiovascular events, including the risk of myocardial infarction. Circulation 2017, 135, 2085–2087. [Google Scholar] [CrossRef]
- Strauss, M.H.; Hall, A.S. Angiotensin receptor blockers do not reduce risk of myocardial infarction, cardiovascular death, or total mortality: Further evidence for the ARB-MI paradox. Circulation 2017, 135, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, C.; Fox, K.M.; Remme, W.J.; Simoons, M.L.; Bertrand, M.; Parrinello, G.; Kluft, C.; Blann, A.; Cokkinos, D.; Ferrari, R.; et al. ACE inhibition with perindopril and endothelial function. Results of a substudy of the EUROPA study: PERTINENT. Cardiovasc. Res. 2007, 73, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnoletti, L.; Bachetti, T.; Bastianon, D.; Francolini, G.; Ferrari, R. Rosuvastatin stimulates eNOS and inhibits apoptosis in HUVECs exposed to sera from cardiovascular diseases patients. J. Mol. Cell. Cardiol. 2007, 42, S226. [Google Scholar] [CrossRef]
- Zacharek, A.; Chen, J.; Cui, X.; Yang, Y.; Chopp, M. Simvastatin increases notch signaling activity and promotes arteriogenesis after stroke. Stroke 2009, 40, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 deficiency and protection against coronary artery disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef] [Green Version]
- Pouwer, M.G.; Pieterman, E.J.; Worms, N.; Keijzer, N.; Jukema, J.W.; Gromada, J.; Gusarova, V.; Princen, H.M.G. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J. Lipid Res. 2020, 61, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.X.; Redon, V.; Yu, H.; Querbes, W.; Pirruccello, J.; Liebow, A.; Deik, A.; Trindade, K.; Wang, X.; Musunuru, K.; et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis 2018, 268, 196–206. [Google Scholar] [CrossRef]
- Camenisch, G.; Pisabarro, M.T.; Sherman, D.; Kowalski, J.; Nagel, M.; Hass, P.; Xie, M.H.; Gurney, A.; Bodary, S.; Liang, X.H.; et al. ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. J. Biol. Chem. 2002, 277, 17281–17290. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Zeng, D.; Shen, Z.; Liao, J.; Wang, X.; Liu, Y.; Zhang, X.; Kong, P. Integrin alphavbeta3 enhances β-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: Implications for microenvironment influence on sorafenib sensitivity. Oncotarget 2016, 7, 40387–40397. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Conner, S.D. Glycogen synthase kinase 3β inhibition enhances Notch1 recycling. Mol. Biol. Cell 2018, 29, 389–395. [Google Scholar] [CrossRef]
- Herrmann, J.; Lerman, L.O.; Mukhopadhyay, D.; Napoli, C.; Lerman, A. Angiogenesis in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1948–1957. [Google Scholar] [CrossRef] [Green Version]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020, 183, 729–737. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Miller, S.E.; Martines, R.B.; Bullock, H.A.; Zaki, S.R. Electron microscopy of SARS-CoV-2: A challenging task. Lancet 2020, 395, e99. [Google Scholar] [CrossRef]
- Dittmayer, C.; Meinhardt, J.; Radbruch, H.; Radke, J.; Heppner, B.I.; Heppner, F.L.; Stenzel, W.; Holland, G.; Laue, M. Why misinterpretation of electron micrographs in SARS-CoV-2-infected tissue goes viral. Lancet 2020, 396, e64–e65. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. 2020, 33, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Ahmetaj-Shala, B.; Peacock, T.P.; Baillon, L.; Swann, O.C.; Gashaw, H.; Barclay, W.S.; Mitchell, J.A. Resistance of endothelial cells to SARS-CoV-2 infection in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells. Front. Cell Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Rauch, A.; Dupont, A.; Goutay, J.; Caplan, M.; Staessens, S.; Moussa, M.; Jeanpierre, E.; Corseaux, D.; Lefevre, G.; Lassalle, F.; et al. Endotheliopathy is induced by plasma from critically ill patients and associated with organ failure in severe COVID-19. Circulation 2020, 142, 1881–1884. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Campo, G.; Contoli, M.; Fogagnolo, A.; Vieceli Dalla Sega, F.; Zucchetti, O.; Ronzoni, L.; Verri, M.; Fortini, F.; Pavasini, R.; Morandi, L.; et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-associated respiratory failure. Platelets. 2021, 32, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The pathophysiological role of neutrophil extracellular traps in inflammatory diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.; Rauch, A.; Staessens, S.; Moussa, M.; Rosa, M.; Corseaux, D.; Jeanpierre, E.; Goutay, J.; Caplan, M.; Varlet, P.; et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1760–1773. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Fortini, F.; Spadaro, S.; Ronzoni, L.; Zucchetti, O.; Manfrini, M.; Mikus, E.; Fogagnolo, A.; Torsani, F.; Pavasini, R.; et al. Time course of endothelial dysfunction markers and mortality in COVID-19 patients: A pilot study. Clin. Transl. Med. 2021, 11, e283. [Google Scholar] [CrossRef]
- Spadaro, S.; Fogagnolo, A.; Campo, G.; Zucchetti, O.; Verri, M.; Ottaviani, I.; Tunstall, T.; Grasso, S.; Scaramuzzo, V.; Murgolo, F.; et al. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit. Care 2021, 25, 74. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, C.; Burtey, S.; Sabatier, F.; Cauchois, R.; Lano, G.; Abdili, E.; Daviet, F.; Arnaud, L.; Brunet, P.; Hraiech, S.; et al. Circulating Endothelial Cells as a Marker of Endothelial Injury in Severe COVID-19. J. Infect. Dis. 2020, 222, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.; Vieceli Dalla Sega, F.; Pavasini, R.; Aquila, G.; Gallo, F.; Fortini, F.; Tonet, E.; Cimaglia, P.; Del Franco, A.; Pestelli, G.; et al. Biological effects of ticagrelor over clopidogrel in patients with stable coronary artery disease and chronic obstructive pulmonary disease. Thromb. Haemost. 2017, 117, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, S.; Mendes, M.C.; Camargo Martins, A.P.; Borges, N.H.; Godoy, T.M.; Miggiolaro, A.F.R.D.; da Silva Dezidério, F.; Machado-Souza, C.; de Noronha, L. Endothelial Dysfunction and Thrombosis in Patients With COVID-19-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, J.; Chen, H.; Zhuge, Y.; Qian, F.; Zhou, K.; Niu, C.; Wang, F.; Qiu, H.; Wang, Z.; et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death. Dis. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.; Exline, M.; Habyarimana, F.; Gavrilin, M.A.; Baker, P.J.; Masters, S.L.; Wewers, M.D.; Sarkar, A. Microparticulate Caspase 1 Regulates Gasdermin D and Pulmonary Vascular Endothelial Cell Injury. Am. J. Respir. Cell Mol. Biol. 2018, 59, 56–64. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortini, F.; Vieceli Dalla Sega, F.; Marracino, L.; Severi, P.; Rapezzi, C.; Rizzo, P.; Ferrari, R. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines 2021, 9, 997. https://doi.org/10.3390/biomedicines9080997
Fortini F, Vieceli Dalla Sega F, Marracino L, Severi P, Rapezzi C, Rizzo P, Ferrari R. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines. 2021; 9(8):997. https://doi.org/10.3390/biomedicines9080997
Chicago/Turabian StyleFortini, Francesca, Francesco Vieceli Dalla Sega, Luisa Marracino, Paolo Severi, Claudio Rapezzi, Paola Rizzo, and Roberto Ferrari. 2021. "Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape" Biomedicines 9, no. 8: 997. https://doi.org/10.3390/biomedicines9080997
APA StyleFortini, F., Vieceli Dalla Sega, F., Marracino, L., Severi, P., Rapezzi, C., Rizzo, P., & Ferrari, R. (2021). Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines, 9(8), 997. https://doi.org/10.3390/biomedicines9080997