Identifying Downregulation of Autophagy Markers in Kawasaki Disease



Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Real-Time PCR
2.3. Statistical Analysis
3. Results
3.1. Demographic Data
3.2. Differential Counts of Leukocytes and Monocytes among the Control Groups and the Children with KD before and after Treatment
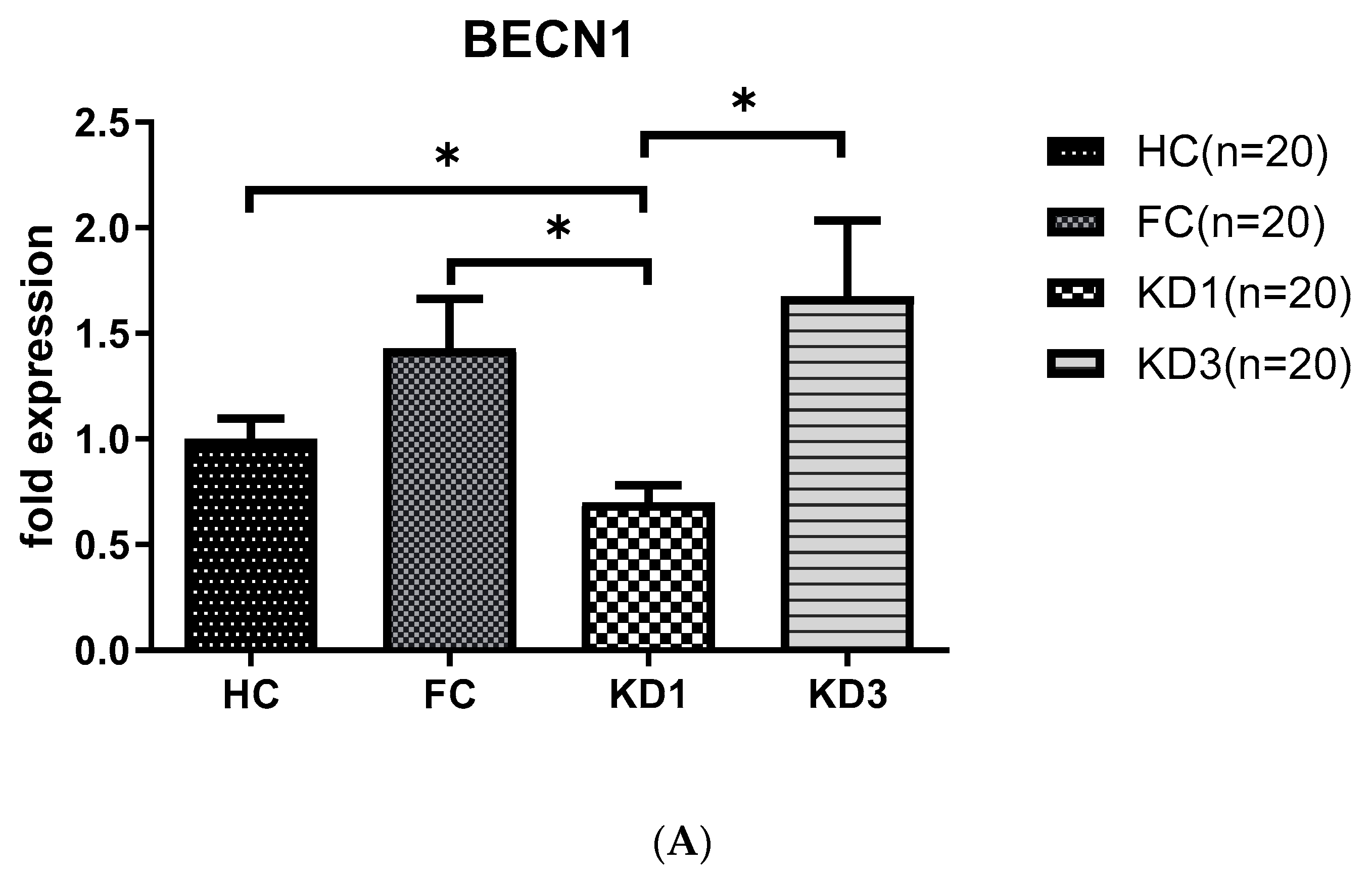
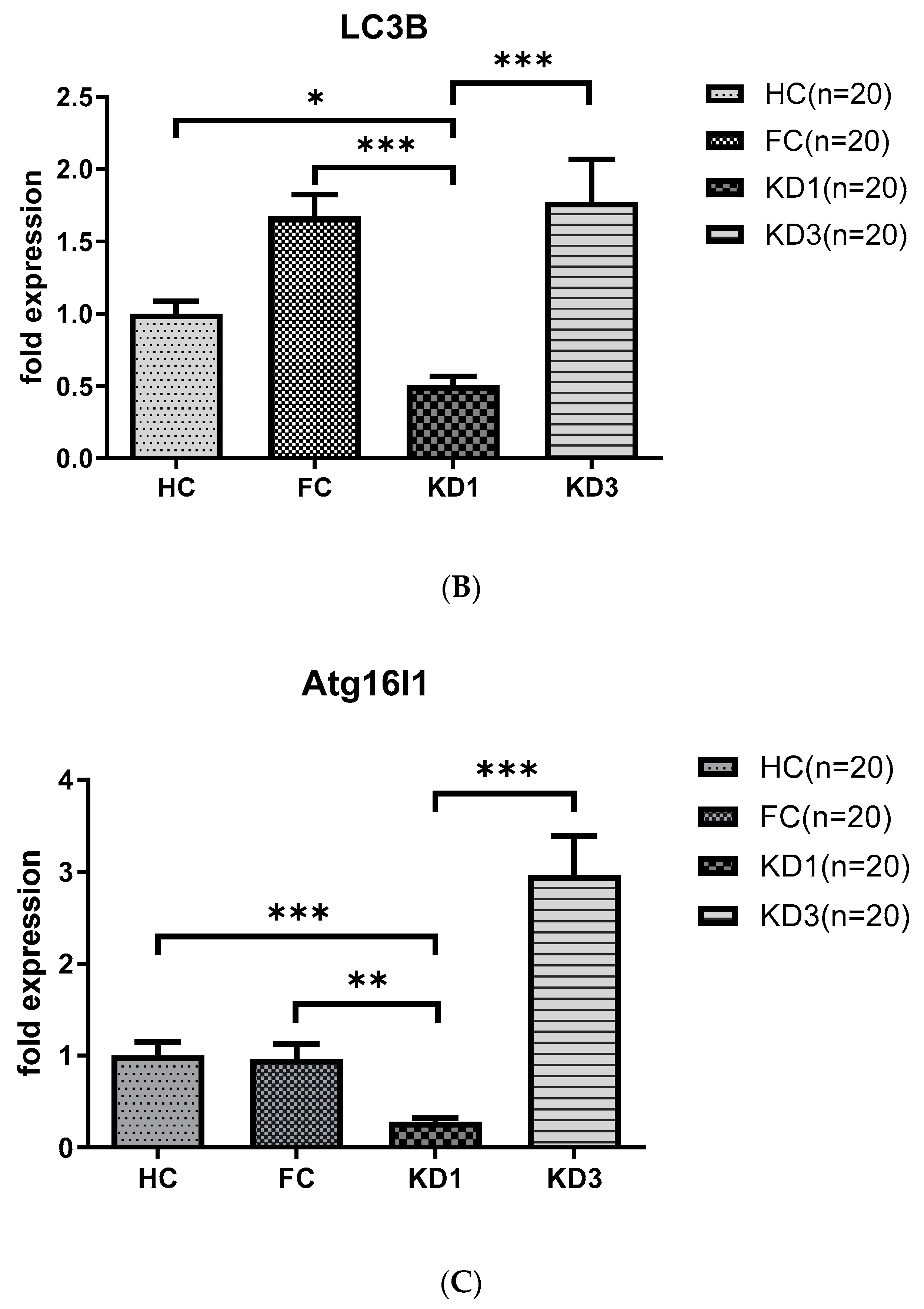
3.3. Analyses of Autophagy Markers mRNA in the Peripheral White Blood Cells of Kawasaki Disease (KD) Patients before and after IVIG Treatment
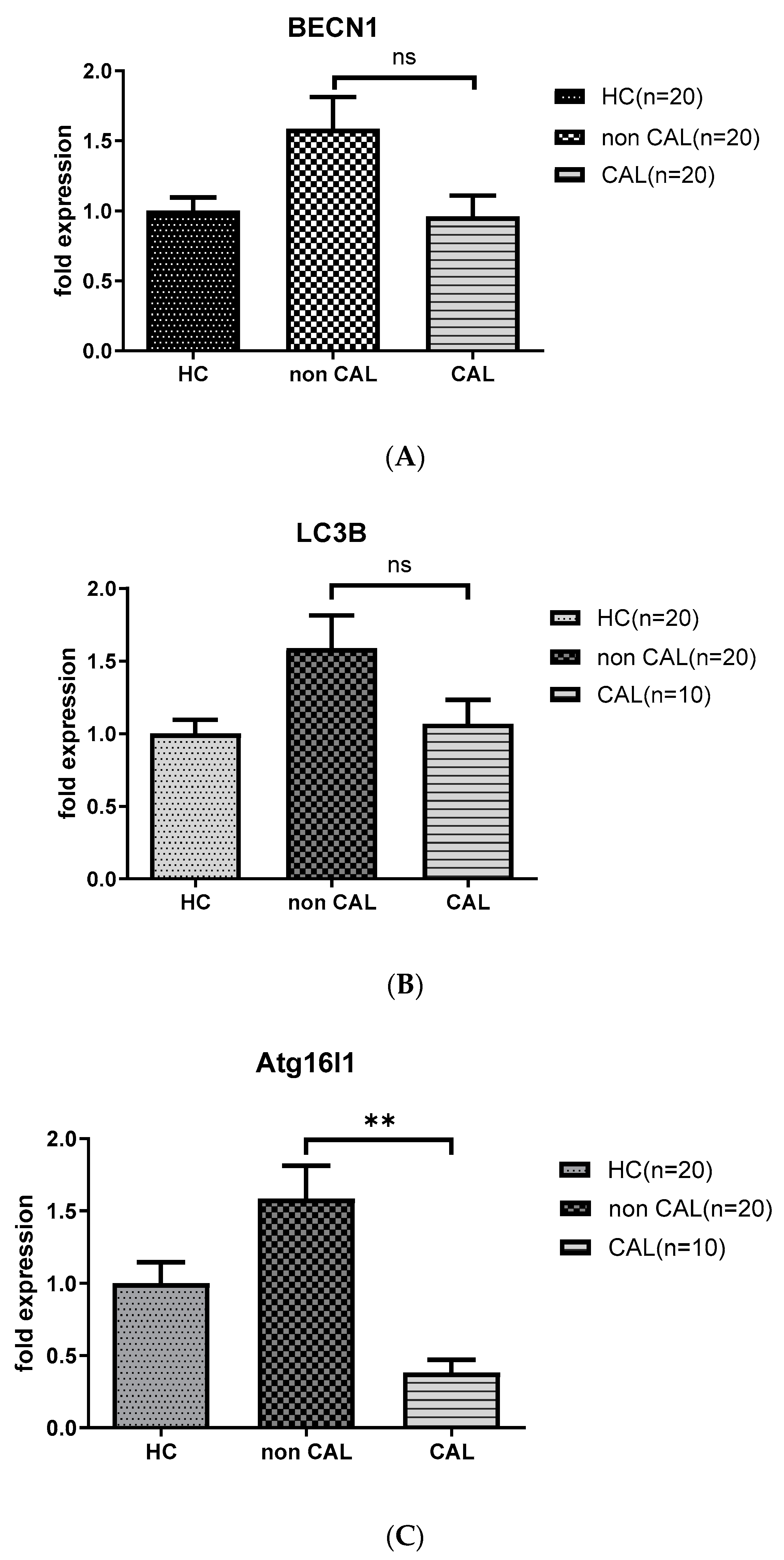
3.4. The Lower Levels of ATG16L1 mRNA Persist in KD Patients with CAL Compared to Those without CAL 21 Days after IVIG Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethics Approval and Consent to Participate
Availability of Data and Materials
References
- Taubert, K.A.; Rowley, A.H.; Shulman, S.T. Nationwide survey of Kawasaki disease and acute rheumatic fever. J. Pediatr. 1991, 119, 279–282. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kosaki, F.; Okawa, S.; Shigematsu, I.; Yanagawa, H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 1974, 54, 271–276. [Google Scholar]
- Suzuki, A.; Kamiya, T.; Kuwahara, N.; Ono, Y.; Kohata, T.; Takahashi, O.; Kimura, K.; Takamiya, M. Coronary arterial lesions of Kawasaki disease: Cardiac catheterization findings of 1100 cases. Pediatr. Cardiol. 1986, 7, 3–9. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals from the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sugimura, T.; Akagi, T.; Sato, N.; Hashino, K.; Maeno, Y.; Kazue, T.; Eto, G.; Yamakawa, R. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 1996, 94, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Newburger, J.W.; Takahashi, M.; Burns, J.C.; Beiser, A.S.; Chung, K.J.; Duffy, C.E.; Glode, M.P.; Mason, W.H.; Reddy, V.; Sanders, S.P.; et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 1986, 315, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Han, R.K.; Silverman, E.D.; Newman, A.; McCrindle, B.W. Management and outcome of persistent or recurrent fever after initial intravenous gamma globulin therapy in acute Kawasaki disease. Arch. Pediatr. Adolesc. Med. 2000, 154, 694–699. [Google Scholar] [CrossRef]
- Hill, J.A. Autophagy in cardiac plasticity and disease. Pediatr. Cardiol. 2011, 32, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Sawada, K.; Shimomura, H.; Kawamura, K.; James, T.N. On the nature of cell death during remodeling of hypertrophied human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 161–175. [Google Scholar] [CrossRef]
- Miyata, S.; Takemura, G.; Kawase, Y.; Li, Y.; Okada, H.; Maruyama, R.; Ushikoshi, H.; Esaki, M.; Kanamori, H.; Li, L.; et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am. J. Pathol. 2006, 168, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.-P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.-P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Saji, T.; Otani, T.; Takeuchi, K.; Nakamura, T.; Arakawa, H.; Kato, T.; Hara, T.; Hamaoka, K.; Ogawa, S.; et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): A randomised, open-label, blinded-endpoints trial. Lancet 2012, 379, 1613–1620. [Google Scholar] [CrossRef]
- Guo, M.M.; Tseng, W.N.; Ko, C.H.; Pan, H.-M.; Hsieh, K.-S.; Kuo, H.-C. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy 2015, 70, 310–318. [Google Scholar] [CrossRef]
- Knaapen, M.W.; Davies, M.J.; De Bie, M.; Haven, A.J.; Martinet, W.; Kockx, M.M. Apoptotic versus autophagic cell death in heart failure. Cardiovasc. Res. 2001, 51, 304–312. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444. [Google Scholar]
- Takahashi, K.; Oharaseki, T.; Yokouchi, Y.; Naoe, S.; Saji, T. Kawasaki disease: Basic and pathological findings. Clin. Exp. Nephrol. 2013, 17, 690–693. [Google Scholar] [CrossRef]
- Rowley, A.H. The etiology of Kawasaki disease: Superantigen or conventional antigen? Pediatr. Infect. Dis. J. 1999, 18, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Meissner, H.C.; Leung, D.Y. Superantigens, conventional antigens and the etiology of Kawasaki syndrome. Pediatr. Infect. Dis. J. 2000, 19, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Matsutani, T.; Toyosaki-Maeda, T.; Suzuki, H.; Uemura, S.; Suzuki, R.; Koike, M.; Hinuma, Y. Relation of streptococcal pyrogenic exotoxin C as a causative superantigen for Kawasaki disease. Pediatr. Res. 2003, 53, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.Y.; Chang, I.S.; Lu, C.Y.; Chiang, B.-L.; Lee, C.-Y.; Chen, P.-J.; Wang, J.-T.; Ho, H.-N.; Chen, D.; Huang, L.-M.; et al. Epidemiologic features of Kawasaki disease in Taiwan, 1996–2002. Pediatrics 2004, 114, e678–e682. [Google Scholar] [CrossRef] [Green Version]
- Cimaz, R.; Falcini, F. An update on Kawasaki disease. Autoimmun. Rev. 2003, 2, 258–263. [Google Scholar] [CrossRef]
- Leung, D.Y.; Meissner, H.C.; Shulman, S.T.; Mason, W.H.; Gerber, M.A.; Glodé, M.P.; Myones, B.L.; Wheeler, J.; Ruthazer, R.; Schlievert, P.M. Prevalence of superantigen-secreting bacteria in patients with Kawasaki disease. J. Pediatr. 2002, 140, 742–746. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.-C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [Green Version]
- Itoh, H.; Matsuo, H.; Kitamura, N.; Yamamoto, S.; Higuchi, T.; Takematsu, H.; Kamikubo, Y.; Kondo, T.; Yamashita, K.; Sasada, M.; et al. Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. J. Leukoc. Biol. 2015, 98, 107–117. [Google Scholar] [CrossRef]
- Matsuo, H.; Itoh, H.; Kitamura, N.; Kamikubo, Y.; Higuchi, T.; Shiga, S.; Ichiyama, S.; Kondo, T.; Takaori-Kondo, A.; Adachi, S. Intravenous immunoglobulin enhances the killing activity and autophagy of neutrophils isolated from immunocompromised patients against multidrug-resistant bacteria. Biochem. Biophys. Res. Commun. 2015, 464, 94–99. [Google Scholar] [CrossRef]
- Newburger, J.W.; Takahashi, M.; Beiser, A.S.; Burns, J.C.; Bastian, J.; Chung, K.J.; Colan, S.D.; Duffy, C.E.; Fulton, D.R.; Glode, M.P.; et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N. Engl. J. Med. 1991, 324, 1633–1639. [Google Scholar] [CrossRef]
- Wallace, C.A.; French, J.W.; Kahn, S.J.; Sherry, D.D. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics 2000, 105, E78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedly, L.A.; Tonnesen, M.G.; Sandhaus, R.A.; Haslett, C.; Guthrie, L.A.; Johnston, R.B.; Henson, P.M.; Worthen, G.S. Neutrophil-mediated injury to endothelial cells. Enhancement by endotoxin and essential role of neutrophil elastase. J. Clin. Investig. 1986, 77, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, H.; Takeshita, S.; Nakatani, K.; Kawamura, Y.; Tokutomi, T.; Sekine, I. Delayed apoptosis of circulating neutrophils in Kawasaki disease. Clin. Exp. Immunol. 2001, 126, 355–364. [Google Scholar] [CrossRef]
- Biezeveld, M.H.; van Mierlo, G.; Lutter, R.; Kuipers, I.M.; Dekker, T.; Hack, C.E.; Newburger, J.W.; Kuijpers, T.W. Sustained activation of neutrophils in the course of Kawasaki disease: An association with matrix metalloproteinases. Clin. Exp. Immunol. 2005, 141, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Mihalache, C.C.; Simon, H.U. Autophagy regulation in macrophages and neutrophils. Exp. Cell Res. 2012, 318, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, G.; Zhao, E.; Liu, K.; Lin, Y.; Tesfa, L.; Tanaka, K.E.; Czaja, M.J. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J. Hepatol. 2016, 64, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, C.P.; Salo, E.; Pelkonen, P. Circulating interleukin-1 beta in patients with Kawasaki disease. N. Engl. J. Med. 1988, 319, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Popper, S.J.; Shimizu, C.; Shike, H.; Kanegaye, J.T.; Newburger, J.W.; Sundel, R.P.; Brown, P.O.; Burns, J.C.; Relman, D.A. Gene-expression patterns reveal underlying biological processes in Kawasaki disease. Genome Biol. 2007, 8, R261. [Google Scholar] [CrossRef] [Green Version]
- Weng, K.P.; Hsieh, K.S.; Ho, T.Y.; Huang, S.-H.; Lai, C.-R.; Chiu, Y.-T.; Lin, C.-C.; Hwang, Y.-T.; Ger, L.-P.; Huang, S.-C. IL-1B polymorphism in association with initial intravenous immunoglobulin treatment failure in Taiwanese children with Kawasaki disease. Circ. J. 2010, 74, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; Van De Veerdonk, F.L.; De Jong, D.J.; Philpott, D.J.; Van Der Meer, J.W.M.; Girardin, S.E.; Joosten, L.A.B.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Hsieh, K.S.; Ming-Huey Guo, M.; Weng, K.-P.; Ger, L.-P.; Chan, W.-C.; Li, S.-C. Next-generation sequencing identifies micro-RNA-based biomarker panel for Kawasaki disease. J. Allergy Clin. Immunol. 2016, 138, 1227–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Z.; Wu, F.; Dong, F.; Chuang, A.Y.; Messer, J.S.; Boone, D.L.; Kwon, J.H. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy 2014, 10, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Chen, J.; Xu, H.G.; Zhou, X.; He, Q.; Li, Y.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.; et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Nakaoka, H.; Takasaki, I.; Hirono, K.; Yamamoto, S.; Kinoshita, K.; Miyao, N.; Ibuki, K.; Ozawa, S.; Watanabe, K.; et al. MicroRNA-93 may control vascular endothelial growth factor A in circulating peripheral blood mononuclear cells in acute Kawasaki disease. Pediatr. Res. 2016, 80, 425–432. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-DeLeon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K.; et al. M2 Macrophage Polarization Mediates Anti-inflammatory Effects of Endothelial Nitric Oxide Signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Karnam, A.; Stephen-Victor, E.; Gilardin, L.; Bhatt, B.; Sharma, V.K.; Rambabu, N.; Patil, V.; Lecerf, M.; Käsermann, F.; et al. Intravenous immunoglobulin mediates anti-inflammatory effects in peripheral blood mononuclear cells by inducing autophagy. Cell Death Dis. 2020, 11, 50. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Autophagy: A new concept in autoimmunity regulation and a novel therapeutic option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HC (n = 20) | FC (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.49 ± 0.78 | 0.932 |
| sex | ♂(14)/♀(6) | ♂(12)/♀(8) | 0.519 |
| WBC | 9.035 ± 2.06 | 9.95 ± 3.47 | 0.371 |
| RBC | 4.81 ± 0.55 | 4.48 ± 0.24 * | 0.033 |
| Hemoglobin | 12.32 ± 0.9 | 11.58 ± 0.94 * | 0.047 |
| Hematocrit | 35.87 ± 2.57 | 35.01 ± 2.59 | 0.397 |
| Platelets | 314.25 ± 74.35 | 361.6 ± 145.1 | 0.243 |
| Segment | 30.75 ± 11.1 | 39.05 ± 22.12 | 0.287 |
| Lymphocyte | 58.17 ± 12.12 | 48.2 ± 21.08 | 0.110 |
| Monocyte | 6.18 ± 2.29 | 8.15 ± 4.31 | 0.202 |
| Eosinophil | 3.41 ± 2.07 | 1.42 ± 1.68 * | 0.014 |
| Basophil | 0.49 ± 0.77 | 0.18 ± 0.34 | 0.239 |
| HC (n = 20) | KD1 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.40 ± 0.69 | 0.700 |
| sex | ♂(14)/♀(6) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.03 ± 2.06 | 13.91 ± 4.98 * | 0.002 |
| RBC | 4.81 ± 0.55 | 4.30 ± 0.60 * | 0.014 |
| Hemoglobin | 12.32 ± 0.9 | 10.76 ± 0.94 *** | 0.000 |
| Hematocrit | 35.87 ± 2.57 | 32.5 ± 2.50 *** | 0.000 |
| Platelets | 314.25 ± 74.35 | 317.93 ± 94.42 | 0.896 |
| Segment | 30.75 ± 11.19 | 59.79 ± 10.40 *** | 0.000 |
| Lymphocyte | 58.17 ± 12.12 | 30.00 ± 9.84 *** | 0.000 |
| Monocyte | 6.18 ± 2.29 | 6.28 ± 2.77 | 0.899 |
| Eosinophil | 3.41 ± 2.07 | 2.42 ± 1.76 | 0.139 |
| Basophil | 0.49 ± 0.77 | 0.15 ± 0.26 | 0.078 |
| FC (n = 20) | KD1 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.49 ± 0.78 | 1.40 ± 0.69 | 0.725 |
| sex | ♂(12)/♀(8) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.95 ± 3.47 | 13.91 ± 4.98 * | 0.038 |
| RBC | 4.48 ± 0.24 | 4.30 ± 0.61 | 0.322 |
| Hemoglobin | 11.58 ± 0.94 | 10.83 ± 0.96 | 0.062 |
| Hematocrit | 35.01 ± 2.59 | 32.5 ± 2.50 * | 0.022 |
| Platelets | 361.60 ± 145.10 | 317.93 ± 94.42 | 0.360 |
| Segment | 39.05 ± 22.12 | 59.79 ± 10.4 * | 0.017 |
| Lymphocyte | 48.2 ± 21.08 | 30.00 ± 9.84 * | 0.026 |
| Monocyte | 8.15 ± 4.31 | 6.28 ± 2.77 | 0.191 |
| Eosinophil | 1.42 ± 1.68 | 2.42 ± 1.76 | 0.163 |
| Basophil | 0.18 ± 0.34 | 0.15 ± 0.26 | 0.804 |
| HC (n = 20) | KD3 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.40 ± 0.69 | 0.75 |
| sex | ♂(14)/♀(6) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.03 ± 2.06 | 8.28 ± 2.13 | 0.291 |
| RBC | 4.81 ± 0.55 | 4.53 ± 0.42 | 0.107 |
| Hemoglobin | 12.32 ± 0.9 | 11.66 ± 0.89 | 0.037 |
| Hematocrit | 35.87 ± 2.57 | 34.86 ± 1.97 | 0.208 |
| Platelets | 314.25 ± 74.35 | 337.64 ± 72.57 | 0.342 |
| Segment | 30.75 ± 11.19 | 29.43 ± 7.57 | 0.691 |
| Lymphocyte | 58.17 ± 12.12 | 61.89 ± 7.52 | 0.267 |
| Monocyte | 6.18 ± 2.29 | 4.90 ± 1.13 * | 0.045 |
| Eosinophil | 3.41 ± 2.07 | 3.26 ± 1.15 | 0.797 |
| Basophil | 0.49 ± 0.77 | 0.32 ± 0.27 | 0.421 |
| FC (n = 20) | KD3 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.49 ± 0.78 | 1.40 ± 0.69 | 0.775 |
| sex | ♂(12)/♀(8) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.95 ± 3.47 | 8.28 ± 2.13 | 0.140 |
| RBC | 4.48 ± 0.24 | 4.53 ± 0.42 | 0.734 |
| Hemoglobin | 11.58 ± 0.94 | 11.66 ± 0.89 | 0.828 |
| Hematocrit | 35.01 ± 2.59 | 34.86 ± 1.97 | 0.876 |
| Platelets | 361.60 ± 145.10 | 338.87 ± 74.89 | 0.594 |
| Segment | 39.05 ± 22.12 | 29.43 ± 7.57 | 0.213 |
| Lymphocyte | 48.20 ± 21.08 | 61.89 ± 7.52 | 0.075 |
| Monocyte | 8.15 ± 4.31 | 4.94 ± 1.09 ** | 0.007 |
| Eosinophil | 1.42 ± 1.68 | 3.26 ± 1.15 ** | 0.003 |
| Basophil | 0.18 ± 0.34 | 0.32 ± 0.27 | 0.245 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.-C.; Huang, Y.-H.; Kuo, H.-C.; Li, S.-C. Identifying Downregulation of Autophagy Markers in Kawasaki Disease. Children 2020, 7, 166. https://doi.org/10.3390/children7100166
Huang F-C, Huang Y-H, Kuo H-C, Li S-C. Identifying Downregulation of Autophagy Markers in Kawasaki Disease. Children. 2020; 7(10):166. https://doi.org/10.3390/children7100166
Chicago/Turabian StyleHuang, Fu-Chen, Ying-Hsien Huang, Ho-Chang Kuo, and Sung-Chou Li. 2020. "Identifying Downregulation of Autophagy Markers in Kawasaki Disease" Children 7, no. 10: 166. https://doi.org/10.3390/children7100166
APA StyleHuang, F. -C., Huang, Y. -H., Kuo, H. -C., & Li, S. -C. (2020). Identifying Downregulation of Autophagy Markers in Kawasaki Disease. Children, 7(10), 166. https://doi.org/10.3390/children7100166