1. Introduction
Niemann–Pick disease (NPD) is a heterogeneous group of lysosomal storage disorders inherited in an autosomal recessive manner [
1]. Niemann–Pick disease is classified based on its genetic and clinical features; there are four types, namely, type A, type B, type C1, and type C2. The affected gene in type A and type B is
SMPD1, which causes an enzymatic deficiency of sphingomyelinase (SMA) [
2]. Niemann–Pick disease type A usually presents with neurological manifestations leading to death in infancy. Niemann–Pick disease type B is the predominant type presenting with visceral enlargement and often surviving into adulthood. Niemann–Pick disease type C is a fatal condition due to mutations in either the
NPC1 or
NPC2 gene. Defects in the NPC1 gene account for approximately 95% of patients, but NPC2 is the cause in approximately 5% of reported cases [
3,
4,
5].
Niemann–Pick disease is caused by the impaired trafficking of unesterified cholesterol and glycolipids in lysosomes and late endosomes, leading to the disturbed transport of various intracellular lipids, the reduced degradation of glucosylceramide, and the lysosomal storage of cholesterol and glycosphingolipids [
6]. Lipid accumulation predominantly in the brain leads to functional and structural brain damage, visceral organ involvement (prolonged neonatal jaundice and hepatosplenomegaly), and pulmonary infiltrates [
7,
8,
9,
10]. This article highlights the diagnostic challenges, and the limited therapeutic options, for a boy with NPC2 disease and the impact of the disease on the family and future pregnancies.
2. Case Report
A 3-year-old Pakistani boy was admitted to our institute with an exacerbation of chronic lung disease. He was born at full term by spontaneous vaginal delivery with a weight of 3.0 kg, and there were no antenatal concerns. The parents are first cousins with no family history of infant death, developmental delay, or genetic disorder. The neonatal period was marked by jaundice, requiring a 4-day course of phototherapy. Later, in the neonatal period, he developed prolonged jaundice with cholestasis characterized by mixed hyperbilirubinemia, elevated liver enzymes, and hepatomegaly.
In the first year of life, he had frequent epistaxis, for which he used to receive olive oil in the nostrils, aiming to control the bleeding and nasal dryness. In the second year, the patient had a mild course of COVID-19 infection that did not require hospital admission. During his second year (a year ago), he also developed frequent respiratory symptoms that required admission to the ward on many occasions and responded to antibiotics, inhaled steroids, and bronchodilators.
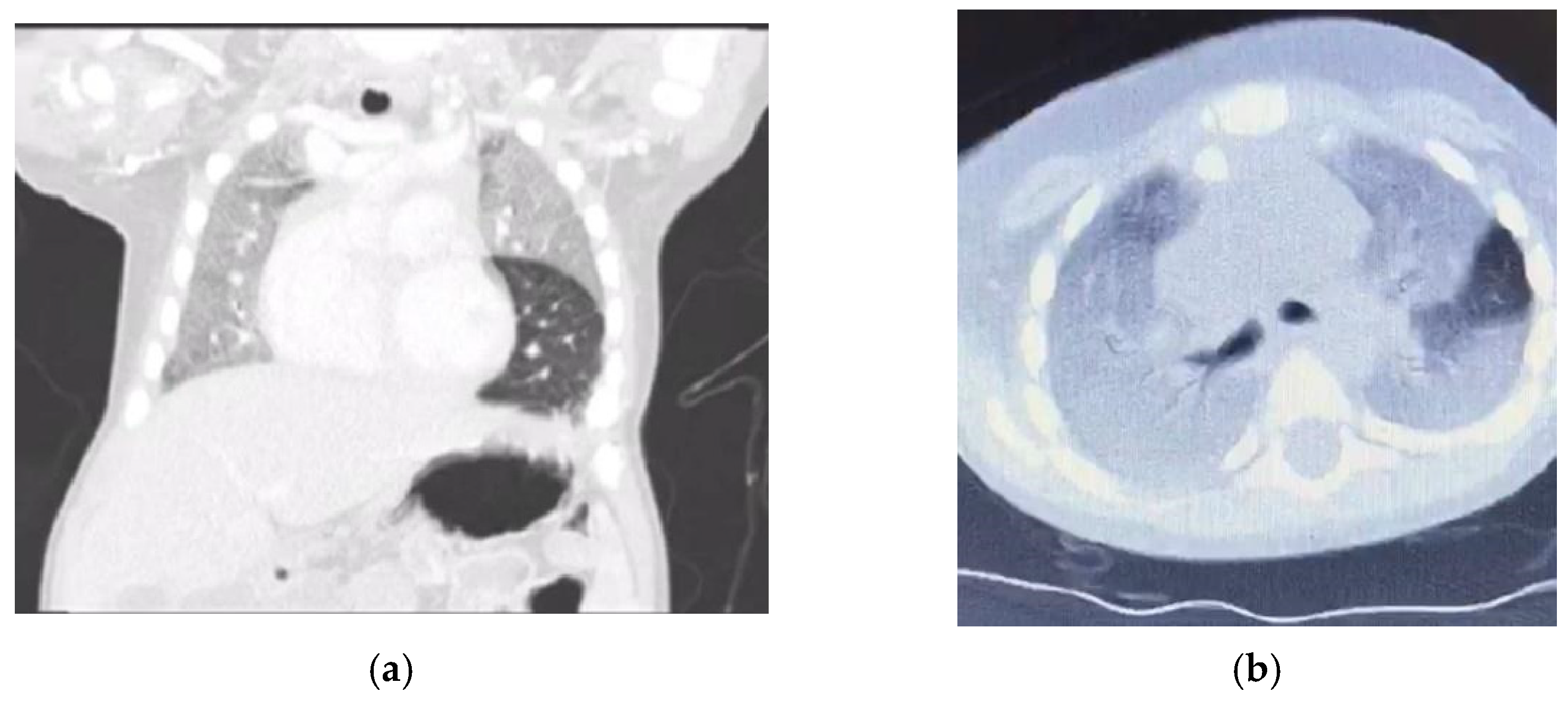
Six months ago, he presented with cough, dyspnoea, and desaturation, requiring admission to the pediatric intensive care unit (PICU) and ventilation for 2 days. Computerized tomography (CT) of the chest (
Figure 1) showed diffuse homogenous ground glass opacity with interstitial septal thickening, which was interpreted by the primary physician as a picture of childhood interstitial lung disease (ChILD) and there was a subsequent prescription of steroids and hydroxychloroquine. He partially responded to treatment and was later discharged home on oxygen by nasal cannula.
In his recent admission to our institute, at the age of three, he had another respiratory exacerbation requiring 5 L/m oxygen. On examination, he was in respiratory distress with subcostal and intercostal recessions, and his respiratory rate was 50 breaths per minute. His oxygen saturation was 70% in room air. He required 5–6 L/m oxygen through the nasal cannula to maintain saturation above 90%. He was febrile, had a heart rate of 140 beats per minute, and had blood pressure of 110/75 mmHg. His weight, length, and head circumference were between the 25th and 50th percentiles. He had grade three clubbing in both hands. Respiratory examination revealed markedly reduced breath sounds at the lung bases with fine inspiratory crackles and absent bronchial breath sounds. His splenic tip was palpable, and his liver was palpable at 4 cm below the costal margin. Neurological assessment revealed global developmental delay with central hypotonia and normal reflexes. Fundoscopy was normal.
3. Results
3.1. Chest X-ray
There were homogenous ground glass opacities involving the entire right lung and the left upper and middle zone with mesocardium due to mild rotation to the right side; there was some retrocardiac collapse and secondary hyperinflation of the left lower lobe but there was no attenuation of the blood vessels to suspect pulmonary emphysema as a possible sign of NPC2 (
Figure 2).
3.2. Blood Work
The blood work, including normal complete blood count, liver, and renal function, and coagulation profile, was benign. On 2 L nasal cannula oxygen, the venous blood gas, consistent with type 2 respiratory failure, showed pH 7.42, PCO2 49 mmHg, P02: 35.8%, O2 saturation 69.9%, HCO3 27.3 mmol/L, base excess 4.6, and lactate 2.3 mmol/L. Abdominal ultrasound revealed moderate hepatosplenomegaly. Whole-exome sequencing (WES) confirmed a homozygous nonsense pathogenic variant in NPC2 (NM_006432.3: c.141C>A), p. (Cys47*), which was previously prescribed as disease-causing by Chikh et al., 2005 (PMID: 15937921). Additionally, the biomarker lyso-SM509 measured by liquid chromatography—mass spectrometry was significantly elevated: 1.6 ng/mL (≤0.9 ng/mL), in the variant coordinates.
3.3. Others
The bronchoalveolar lavage (
Figure 3) was turbid and slightly frothy, and the sedimentation materials in the lower part of the container were suggestive of alveolar proteinosis. They showed numerous macrophages, and the periodic acid-Schiff staining was positive, while the Gram staining and culture were negative. Unfortunately, lipid-laden macrophage, triglyceride, and cholesterol tests were not performed.
3.4. Management
Upon admission to our hospital, the working diagnosis was the exacerbation of the interstitial lung disease of childhood (ChILD) with possible lipoid pneumonia based on the clear history of the instillation of olive oil in the nostrils for several months. However, the severity of the lung involvement together with the presence of neonatal jaundice and hepatosplenomegaly raised the possibility of lipid storage disease (Niemann–Pick or Gaucher disease). Awaiting the result of the WES, the patient received supportive therapy, including noninvasive ventilation, a course of intravenous Tazocin, proton pump inhibitor, and hydration therapy with salbutamol, hypertonic saline, and frequent chest physiotherapy. No clinical improvement was observed, and he remained markedly hypoxic despite two weeks of supportive therapy. Thereafter, therapeutic lung lavage was performed. Unfortunately, this procedure was not tolerated well, with a worsening of his respiratory status that required high-frequency oscillatory ventilation (HFOV) for ten more days and then he was switched to conventional mechanical ventilation and remained on this ventilatory mode. He received low-fat feed with a medium-chain triglyceride (MCT) supplement given through a nasogastric tube. As the therapeutic options were limited and the prognosis was poor, the family received genetic counseling and a do not resuscitate code was discussed, unfortunately the patient passed away.
4. Discussion
Niemann–Pick disease type C2 is a rare lipid storage disorder characterized by a slowly progressive course leading to death in infancy. Its main clinical features include cholestasis, hepatosplenomegaly, and pulmonary and neurological insult (9, 10). Similar to our patient who presented with cholestasis, Vanier et al. described a child with NPC2 who presented with prolonged jaundice and cholestasis, which was misdiagnosed as galactosemia, and other cases in the literature were misdiagnosed with neonatal haemochromatosis [
3]. In our case, presented with jaundice, visceromegaly and early onset of neurological manifestations in the late infantile period, however, the respiratory manifestations were worse compared to published literatures [
3,
4,
7,
8]. A young child with unexplained cholestasis, especially if associated with hepatosplenomegaly and hypotonia, should alert physicians to the possibility of NPD.
Niemann–Pick disease type C2 is predominantly respiratory, in contrast to NPC1 [
7,
8]. There are three types of pulmonary alveolar proteinosis (PAP): autoimmune (the most common), hereditary, and secondary PAP, and coexistence between NPD and PAP is well established in the literature [
9,
10]. The coexistence of NPD and PAP is the main presentation of NPC2, leading to respiratory failure [
11,
12]. Similarly, our patient presented with progressive respiratory insufficiency. Furthermore, the instillation of olive oil in his nostrils with possible lipoid pneumonia possibly worsened the respiratory status of our patient [
13]. Moreover, the chest X-ray showed homogenous opacity involving the entire right lung and the upper and middle zones of the left lung with an air bronchogram, and the chest CT showed a ground glass appearance together with intrapulmonary fat infiltrate with a Hounsfield unit score of −40 HFU (−40 to −130) PAP [
14,
15]. Our case showed ground glass opacities, mild smooth interlobular septal thickening, and intralobular lines as a crazy-paving pattern that could suggest a diagnosis of NPD with no evidence of emphysematous lobe, which has been reported as a sign of NPC2 [
16,
17].
Therapeutic lung lavage is the main supportive therapy in both PAP and lipoid pneumonia, and overlap between the two conditions is common. However, the coexistence of the two conditions has not been reported in the literature to the best of our knowledge. In our experience, gross inspection of the lavage container may help in differentiating between these two conditions. The proteinous material of PAP is likely to sink to the bottom of the container; the color of the lavage is usually pink with a frothy floating substance, as the lipid part will float on the top of the container due to being less dense than normal saline (
Table 1).
No curative therapy is available for NPC2. Multidisciplinary team input would help to support the patient and the family (9, 10). Miglustat is the first and the only approved medication for NPC with neurological manifestations. It is a glucosylceramide synthase inhibitor and an essential enzyme for the synthesis of glycosphingolipids. It has been shown to increase the survival of patients with NPD by five years from the date of diagnosis or approximately ten years from the onset of neurologic manifestations [
9,
18,
19]. There are limited data on other possible therapeutic options, such as stem cell transplantation and liver and lung transplantation. In the current era of genetics, gene therapy may well be an option in the future [
20,
21,
22,
23,
24,
25]. Preventive measures, including antenatal diagnosis and termination of pregnancy with an affected fetus, are viable options for families with a history of NPC2. Additionally, preimplantation genetic testing is the preferred measure if available [
9].
5. Conclusions
Niemann–Pick disease type C2 is a lethal, progressive, and chronically debilitating disease with multisystem involvement. It starts with neonatal jaundice, with variable degrees of visceromegaly and psychomotor retardation and pulmonary involvement. In this case, NPC2 was misdiagnosed as ChILD, resulting in unnecessary treatment with steroids and hydroxychloroquine, thus highlighting the importance of a detailed history and a vigilant investigation of past illnesses; in this case, discovering the neonatal hepatic manifestations, combined with respiratory and neurological involvement suggested the correct diagnosis. Lipoid pneumonia should be considered as an important differential diagnosis, especially in higher incidence areas such as Saudi Arabia. Lavage is very beneficial to differentiate alveolar proteinosis from lipoid pneumonia. Whole-exome sequencing is the preferred confirmatory diagnostic method. Currently, there is no cure for NPC2.
Author Contributions
A.A.-S. observed the patient and wrote the manuscript, K.A.-S. and A.S.M. collected the literature, A.B.M. managed the patient in intensive care and S.M. supervised and contributed to manuscript drafting. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
This work was conducted in Riyadh Care Hospital and National Hospital, Riyad, Saudi Arabia. Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Acknowledgments
The authors would like to thank the patient and the parents for agreeing to the publishing of this case.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| NPD | Niemann–Pick disease |
| NPC | Niemann–Pick disease Type C |
| LSD | Lysosomal storagedisorders
|
| IEMs | Inborn error of metabolism
|
| ChILD | Childhood Interstitial Lung Disease |
| CT | Computed Tomography
|
| GERD | Gastroesophageal reflux disease |
| BAL | Broncho-alveolar lavage |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| PAP | Pulmonary Alveolar Proteinosis |
| PAS | Periodic acid-Schiff |
References
- Sideris, A.A.; Josephson, M. Pulmonary alveolar proteinosis and Niemann Pick disease type B: An unexpected combination Georgios. Respir. Med. Case Rep. 2016, 19, 37–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Cheema, H.; Saeed, A.; Anjum, N.; Waheed, N. Phenotypic diversity of Niemann-Pick disease type C in Pakistani children. Mol. Genet. Metab. 2020, 129, S39. [Google Scholar] [CrossRef]
- Verot, L.; Chikh, K.; Freydiere, E.; Honore, R.; Vanier, M.T.; Millat, G. Niemann-Pick C disease: Functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin. Genet. 2007, 71, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Nafissi, S.; Shamshiri, H.; Nejad, M.M.; Elahi, E. Identification of mutation in NPC2 by exome sequencing results in diagnosis of Niemann-Pick disease type C. Mol. Genet. Metab. 2013, 110, 139–144. [Google Scholar] [CrossRef]
- Nicholson, A.G.; Florio, R.; Hansell; du Bois, R.M.; Wells, A.U.; Hughes, P.; Ramadan, H.K.; Mackinlay, C.I.; Brambilla, E.; Ferretti, G.R.; et al. Pulmonary involvement by Niemann–Pick disease. A report of six cases. Histopathology 2006, 48, 481–628. [Google Scholar] [CrossRef]
- Minai, O.A.; Sullivan, E.J.; Stoller, J.K. Pulmonary involvement in Niemann–Pick disease: Case report and literature review. Respir. Med. 2000, 94, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M. Niemann-Pick Disease Type C. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Eds.; Initial Posting January 26, 2000; [Updated December 10, 2020]; Bookshelf; University of Washington: Seattle, WA, USA, 2000; pp. 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books (accessed on 22 November 2022).
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Gene Reviews Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [Green Version]
- Yaman, A.; Eminoğlu, F.T.; Kendirli, T.; Ödek, Ç.; Ceylaner, S.; Kansu, A.; İnce, E.; Deda, G. A rare cause of fatal pulmonary alveolar proteinosis: Niemann-Pick disease type C2 and a novel mutation. J. Pediatr. Endocrinol. Metab. 2015, 28, 1163–1167. [Google Scholar] [CrossRef]
- Bjurulf, B.; Spetalen, S.; Erichsen, A.; Vanier, M.T.; Strøm, E.H.; Strømme, P. Niemann-Pick disease type C2 presenting as fatal pulmonary alveolar lipoproteinosis: Morphological findings in lung and nervous tissue. Med. Sci. Monit. 2008, 14, CS71–CS75. [Google Scholar]
- Papiris, S.A.; Tsirigotis, P.; Kolilekas, L.; Papadaki, G.; Papaioannou, A.; Triantafillidou, C.; Papaporfyriou, A.; Karakatsani, A.; Kagouridis, K.; Griese, M.; et al. Pulmonary alveolar proteinosis: Time to shift. Expert Rev. Respir. Med. 2015, 9, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamrani, A.; Al-Zaid, M.; AlFadl, S. Lipoid pneumonia treated with modified therapeutic lung lavage. Sudan Med. J. 2020, 21, 1–7. [Google Scholar]
- Betancourt, S.L.; Martinez-Jimenez, S.; Rossi, S.E.; Truong, M.T.; Carrillo, J.; Erasmus, J.J. Lipoid Pneumonia: Spectrum of Clinical and Radiologic Manifestations. Am. J. Roentgenol. 2010, 194, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Von Ranke, F.M.; Freitas, H.M.P.; Mançano, A.D.; Rodrigues, R.S.; Hochhegger, B.; Escuissato, D.; Neto, C.A.A.; da Silva, T.K.; Marchiori, E. Pulmonary Involvement in Niemann–Pick Disease: A State-of-the-Art Review. Lung 2016, 194, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Elleder, M.; Houštková, H.; Zeman, J.; Ledvinová, J.; Poupětová, H. Pulmonary storage with emphysema as a sign of Niemann–Pick type C2 disease (second complementation group). Report of a case. Virchows Arch. 2001, 439, 206–211. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Galanaud, D.; Tourbah, A.; Lehéricy, S.; Leveque, N.; Heron, B.; de Villemeur, T.B.; Guffon, N.; Feillet, F.; Baumann, N.; Vanier, M.T.; et al. 24-month treatment with miglustat of three patients with Niemann-Pick disease type C: Follow up using brain spectroscopy. Mol. Genet. Metab. 2009, 96, 55–58. [Google Scholar] [CrossRef]
- Hsu, Y.-S.; Hwu, W.-L.; Huang, S.-F.; Lu, M.-Y.; Chen, R.-L.; Lin, D.-T.; Peng, S.S.F.; Lin, K.-H. Niemann–Pick disease type C (a cellular cholesterol lipidosis) treated by bone marrow transplantation. Bone Marrow Transplant. 1999, 24, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Mora, V.M.; Osorio, J.S.; Iturbe, D.; Tello, S.; Guzmán, Y.; Sánchez, L.; Gómez, J.J.; Cifrián, J.M. Double-Lung Transplantation in a Patient with Pulmonary Type B Niemann-Pick Disease: A Valid Treatment Option. Case Rep. Transplant. 2022, 2022, c5428381. [Google Scholar] [CrossRef]
- Kumagai, T.; Terashima, H.; Uchida, H.; Fukuda, A.; Kasahara, M.; Kosuga, M.; Okuyama, T.; Tsunoda, T.; Inui, A.; Fujisawa, T.; et al. A case of Niemann-Pick disease type C with neonatal liver failure initially diagnosed as neonatal hemochromatosis. Brain Dev. 2019, 41, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, C.; Superina, R.; Mohammad, S. Normal long-term neurologic and graft outcome after liver transplantation in an infant with Neimann-Pick type C disease. Am. J. Transplant. 2021, 22, 646–648. [Google Scholar] [CrossRef]
- Chikh, K.; Rodriguez, C.; Vey, S.; Vanier, M.T.; Millat, G. Niemann-Pick type C disease: Subcellular location and functional characterization of NPC2 proteins with naturally occurring missense mutations. Hum. Mutat. 2005, 26, 20–28. [Google Scholar] [CrossRef]
- Mutation Taster Documentaion. Available online: https://www.mutationtaster.org/info/documentation.html (accessed on 22 November 2022).
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).







{kind=link}
{kind=link}
{kind=link}


