
‘Teratoid’ Hepatoblastoma: An Intriguing Variant of Mixed Epithelial-Mesenchymal Hepatoblastoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Roots of the ‘Teratoid’ Concept
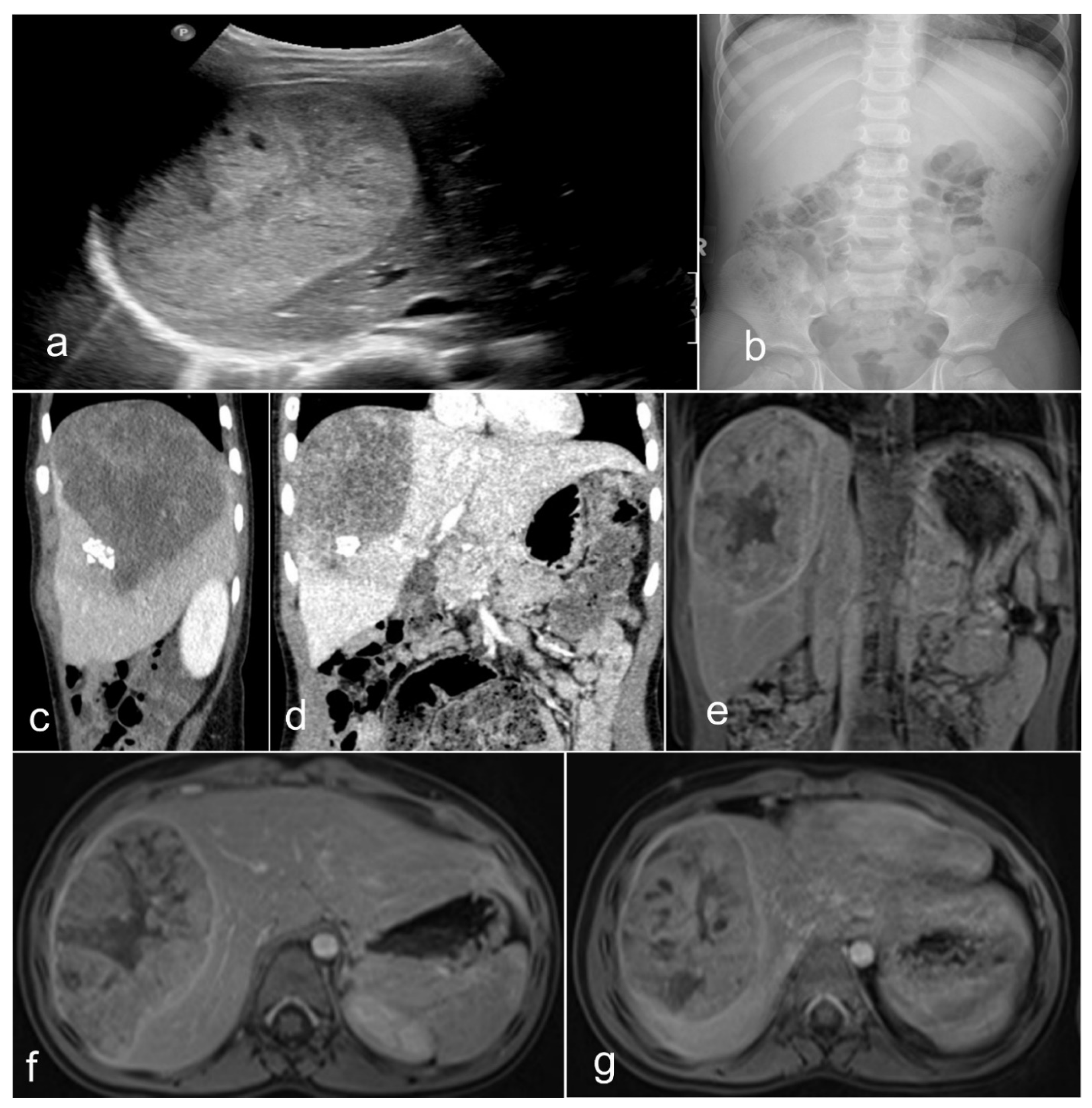
1.2. Clinical-Radiological Features
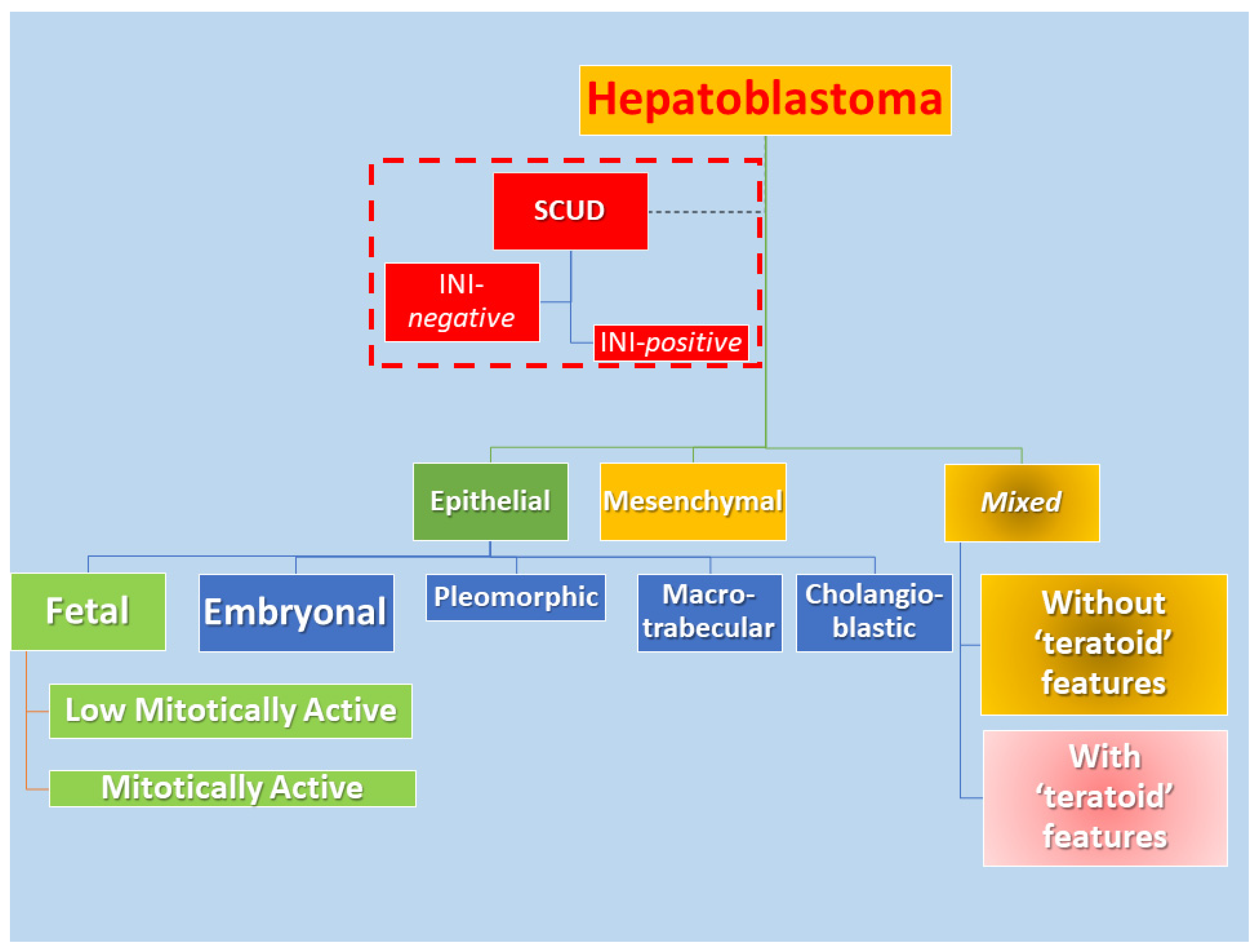
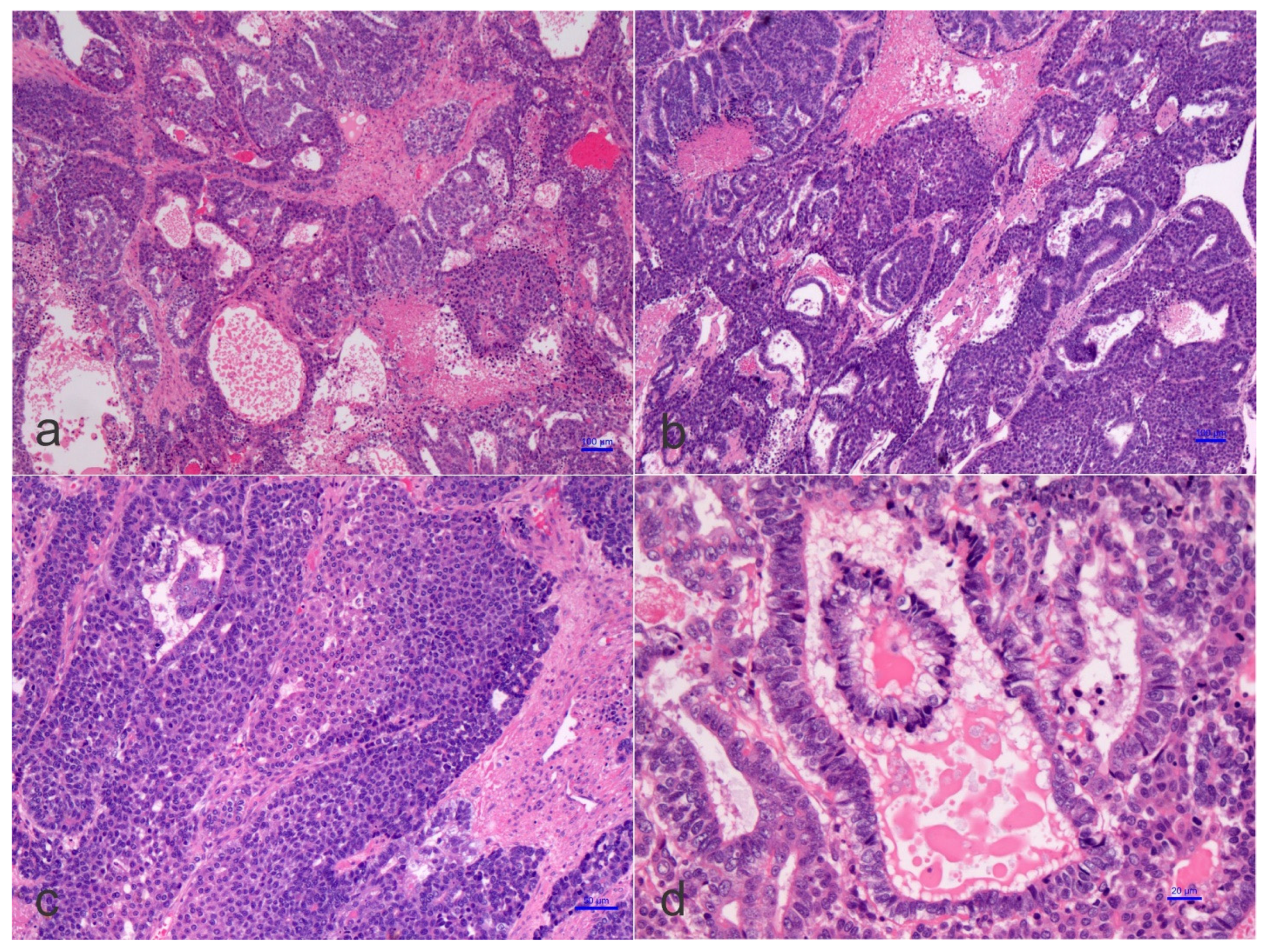
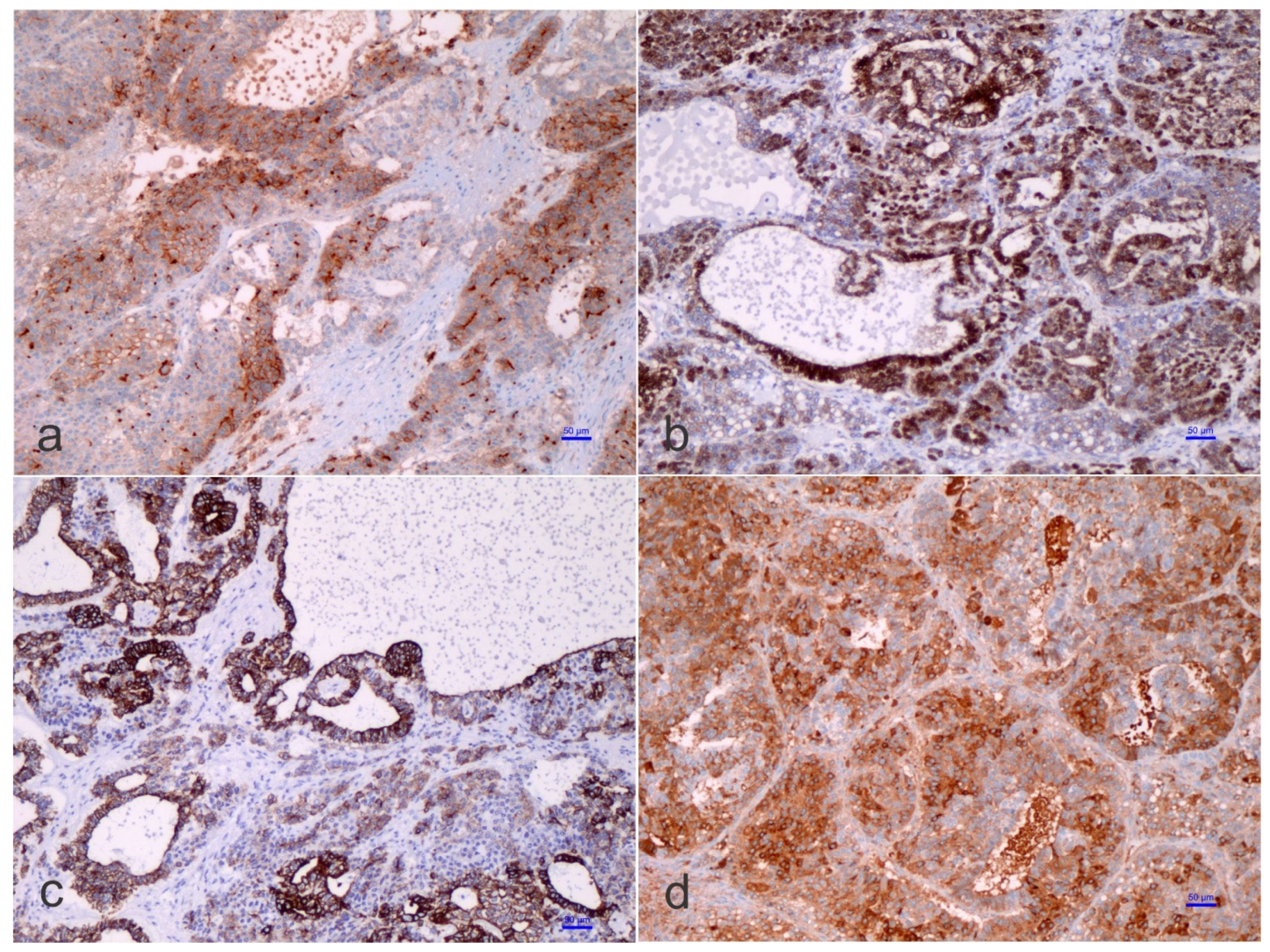
1.3. Pathological Features
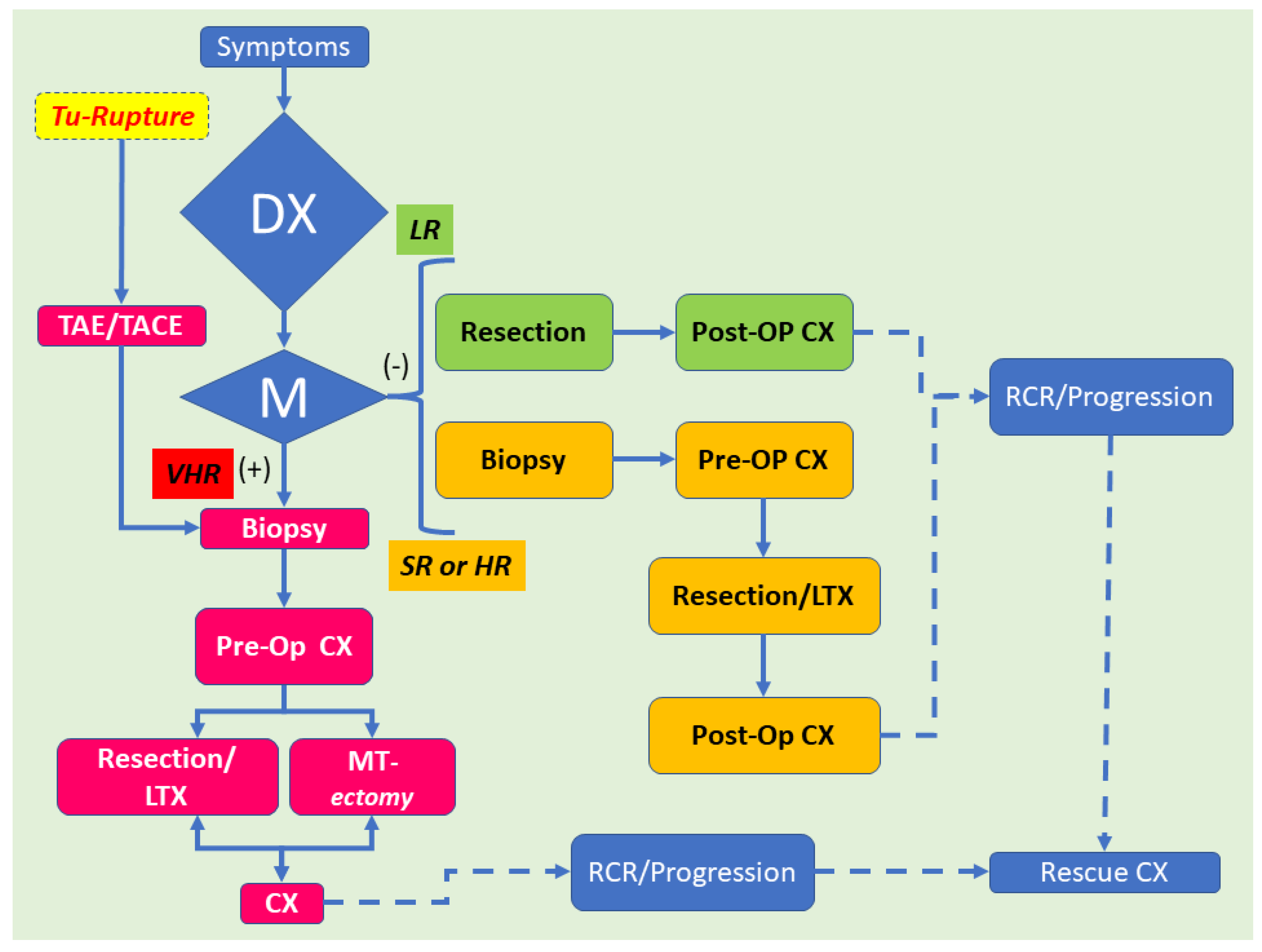
1.4. Management
2. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviation
| AFP | α-fetoprotein |
| BCL-2 | B-cell lymphoma |
| CEA | Carcino-embryonic antigen |
| CK-7 | (Cyto-)Keratin 7 |
| CK-8 | (Cyto-)Keratin 8 |
| CK-18 | (Cyto-)Keratin 18 |
| CK-19 | (Cyto-)Keratin 19 |
| COG | Children’s Oncology Group |
| FNA | Fine Needle Aspiration |
| GPOG | German Pediatric Oncology Group |
| GWAS | Genomic wide association studies |
| HB | Hepatoblastoma |
| IPTA | International Pediatric Transplant Association |
| OV-6 | Oval cells-6 |
| PD-1 | Programmed death 1 |
| PD-L1 | Programmed death-ligand 1 |
| PRETEXT | Pre-treatment extent of disease staging system |
| SCUD | Small cell undifferentiated |
| SIOP | International Society of Pediatric Oncology |
| SIOPEL | International Childhood Liver Tumors Strategy Group |
References
- Sharma, D.; Subbarao, G.; Saxena, R. Hepatoblastoma. Semin. Diagn. Pathol. 2017, 34, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Saxena, R. Primary mesenchymal liver tumors of childhood. Semin. Diagn. Pathol. 2017, 34, 201–207. [Google Scholar] [CrossRef]
- Kromhout, H.; Friesen, M.; Marques, M.M.; Sergi, C.M.; Abdallah, M.; Benke, G.; Cesta, M.; Germolec, D.; Houck, K.; Ichihara, G.; et al. Carcinogenicity of isobutyl nitrite, beta-picoline, and some acrylates. Lancet Oncol. 2018, 19, 1020–1022. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Loomis, D.; Grosse, Y.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Scoccianti, C.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of tetrachlorvinphos, parathion, malathion, diazinon, and glyphosate. Lancet Oncol. 2015, 16, 490–491. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Loomis, D.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of pentachlorophenol and some related compounds. Lancet Oncol. 2016, 17, 1637–1638. [Google Scholar] [CrossRef]
- Grosse, Y.; Loomis, D.; Guyton, K.Z.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Some chemicals that cause tumours of the urinary tract in rodents. Lancet Oncol. 2017, 18, 1003–1004. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zemmel, R. Value of novelty? Nat. Rev. Drug Discov. 2002, 1, 571–572. [Google Scholar] [CrossRef]
- Kingsmore, S.F.; Lindquist, I.E.; Mudge, J.; Gessler, D.D.; Beavis, W.D. Genome-wide association studies: Progress and potential for drug discovery and development. Nat. Rev. Drug Discov. 2008, 7, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.C.; Zeggini, E.; Panoutsopoulou, K. Do genome-wide association scans have potential for translation? Clin. Chem. Lab. Med. 2011, 50, 255–260. [Google Scholar] [CrossRef]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manivel, C.; Wick, M.R.; Abenoza, P.; Dehner, L.P. Teratoid hepatoblastoma. The nosologic dilemma of solid embryonic neoplasms of childhood. Cancer 1986, 57, 2168–2174. [Google Scholar] [CrossRef]
- Misugi, K.; Reiner, C.B. A malignant true teratoma of liver in childhood. Arch. Pathol. 1965, 80, 409–412. [Google Scholar] [PubMed]
- Czauderna, P.; Lopez-Terrada, D.; Hiyama, E.; Haberle, B.; Malogolowkin, M.H.; Meyers, R.L. Hepatoblastoma state of the art: Pathology, genetics, risk stratification, and chemotherapy. Curr. Opin. Pediatr. 2014, 26, 19–28. [Google Scholar] [CrossRef]
- Lopez-Terrada, D.; Alaggio, R.; de Davila, M.T.; Czauderna, P.; Hiyama, E.; Katzenstein, H.; Leuschner, I.; Malogolowkin, M.; Meyers, R.; Ranganathan, S.; et al. Towards an international pediatric liver tumor consensus classification: Proceedings of the Los Angeles COG liver tumors symposium. Mod. Pathol. 2014, 27, 472–491. [Google Scholar] [CrossRef] [Green Version]
- Russo, P. Liver including tumors, gallbladder, and biliary tree. In Potter’s Pathology of the Fetus, Infant and Child, 2nd ed.; Gilbert-Barness, E., Ed.; Mosby-Elsevier: Philadelphia, PA, USA, 2007; pp. 1207–1280. [Google Scholar]
- Honeyman, J.N.; Simon, E.P.; Robine, N.; Chiaroni-Clarke, R.; Darcy, D.G.; Lim, I.I.; Gleason, C.E.; Murphy, J.M.; Rosenberg, B.R.; Teegan, L.; et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 2014, 343, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Sergi, C.M. Hepatocellular Carcinoma, Fibrolamellar Variant: Diagnostic Pathologic Criteria and Molecular Pathology Update. A Primer. Diagnostics 2015, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith-Wiedemann Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; NCBI, University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cohen, M.M., Jr. Beckwith-Wiedemann syndrome: Historical, clinicopathological, and etiopathogenetic perspectives. Pediatr. Dev. Pathol. 2005, 8, 287–304. [Google Scholar] [CrossRef]
- Dasouki, M.; Barr, M., Jr. Trisomy 18 and hepatic neoplasia. Am. J. Med. Genet. 1987, 27, 203–205. [Google Scholar] [CrossRef]
- Mamlok, V.; Nichols, M.; Lockhart, L.; Mamlok, R. Trisomy 18 and hepatoblastoma. Am. J. Med. Genet. 1989, 33, 125–126. [Google Scholar] [CrossRef]
- Maruyama, K.; Ikeda, H.; Koizumi, T. Hepatoblastoma associated with trisomy 18 syndrome: A case report and a review of the literature. Pediatr. Int. 2001, 43, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Schnater, J.M.; Schouten-van Meeteren, A.Y.; Heins, Y.M.; Aronson, D.C. Hepatoblastoma in a patient with a partial trisomy 9p syndrome: A case report. Cancer Genet. Cytogenet. 2005, 156, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Tran, H.C.; Randolph, L.; Mascarenhas, L.; Venkatramani, R. Hepatoblastoma in a 15-month-old female with trisomy 13. Am. J. Med. Genet. A 2014, 164A, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Mugishima, H.; Shichino, H.; Suzuki, T.; Chin, M.; Koshinaga, S.; Inoue, M.; Harada, K. Karyotypic analyses of hepatoblastoma. Report of two cases and review of the literature suggesting chromosomal loci responsible for the pathogenesis of this disease. Cancer Genet. Cytogenet. 1999, 114, 42–50. [Google Scholar] [CrossRef]
- Parada, L.A.; Limon, J.; Iliszko, M.; Czauderna, P.; Gisselsson, D.; Hoglund, M.; Kullendorff, C.M.; Wiebe, T.; Mertens, F.; Johansson, B. Cytogenetics of hepatoblastoma: Further characterization of 1q rearrangements by fluorescence in situ hybridization: An international collaborative study. Med. Pediatr. Oncol. 2000, 34, 165–170. [Google Scholar] [CrossRef]
- Begemann, M.; Trippett, T.M.; Lis, E.; Antunes, N.L. Brain metastases in hepatoblastoma. Pediatr. Neurol. 2004, 30, 295–297. [Google Scholar] [CrossRef]
- Endo, E.G.; Walton, D.S.; Albert, D.M. Neonatal hepatoblastoma metastatic to the choroid and iris. Arch. Ophthalmol. 1996, 114, 757–761. [Google Scholar] [CrossRef]
- Matsunaga, T.; Sasaki, F.; Ohira, M.; Hashizume, K.; Hayashi, A.; Hayashi, Y.; Mugishima, H.; Ohnuma, N.; Japanese Study Group for Pediatric Liver, T. Analysis of treatment outcome for children with recurrent or metastatic hepatoblastoma. Pediatr. Surg. Int. 2003, 19, 142–146. [Google Scholar] [CrossRef]
- Neu, S.M. Hepatoblastoma in an equine fetus. J. Vet. Diagn. Investig. 1993, 5, 634–637. [Google Scholar] [CrossRef] [Green Version]
- Prater, P.E.; Patton, C.S.; Held, J.P. Pleural effusion resulting from malignant hepatoblastoma in a horse. J. Am. Vet. Med. Assoc. 1989, 194, 383–385. [Google Scholar]
- Loynachan, A.T.; Bolin, D.C.; Hong, C.B.; Poonacha, K.B. Three equine cases of mixed hepatoblastoma with teratoid features. Vet. Pathol. 2007, 44, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.; Park, Y.N.; Kim, S.E.; Noh, T.W.; Park, C. Teratoid hepatoblastoma: Multidirectional differentiation of stem cell of the liver. Yonsei Med. J. 2001, 42, 431–435. [Google Scholar] [CrossRef]
- Zimmermann, A. The emerging family of hepatoblastoma tumours: From ontogenesis to oncogenesis. Eur. J. Cancer 2005, 41, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A. Pediatric liver tumors and hepatic ontogenesis: Common and distinctive pathways. Med. Pediatr. Oncol. 2002, 39, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A. Hepatoblastoma with cholangioblastic features (‘cholangioblastic hepatoblastoma’) and other liver tumors with bimodal differentiation in young patients. Med. Pediatr. Oncol. 2002, 39, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Sergi, C.; Adam, S.; Kahl, P.; Otto, H.F. The remodeling of the primitive human biliary system. Early Hum. Dev. 2000, 58, 167–178. [Google Scholar] [CrossRef]
- Sergi, C.; Kahl, P.; Otto, H.F. Contribution of apoptosis and apoptosis-related proteins to the malformation of the primitive intrahepatic biliary system in Meckel syndrome. Am. J. Pathol. 2000, 156, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Sergi, C.; Adam, S.; Kahl, P.; Otto, H.F. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr. Dev. Pathol. 2000, 3, 568–583. [Google Scholar] [CrossRef] [PubMed]
- Fiegel, H.C.; Gluer, S.; Roth, B.; Rischewski, J.; von Schweinitz, D.; Ure, B.; Lambrecht, W.; Kluth, D. Stem-like cells in human hepatoblastoma. J. Histochem. Cytochem. 2004, 52, 1495–1501. [Google Scholar] [CrossRef] [Green Version]
- Variend, S.; Spicer, R.D.; Mackinnon, A.E. Teratoid Wilms’ tumor. Cancer 1984, 53, 1936–1942. [Google Scholar] [CrossRef]
- Zhou, S.; O’Gorman, M.R.; Yang, F.; Andresen, K.; Wang, L. Glypican 3 as a Serum Marker for Hepatoblastoma. Sci. Rep. 2017, 7, 45932. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.L.; Qin, H.; Yan, S.Q.; Zhou, L.S.; Chen, P.; Zhao, D. Expression of glypican-3 is highly associated with pediatric hepatoblastoma: A systemic analysis. Asian Pac. J. Cancer Prev. 2015, 16, 1029–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruck, P.; Xiao, J.C.; Pietsch, T.; Von Schweinitz, D.; Kaiserling, E. Hepatic stem-like cells in hepatoblastoma: Expression of cytokeratin 7, albumin and oval cell associated antigens detected by OV-1 and OV-6. Histopathology 1997, 31, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Haruna, Y.; Saito, K.; Nalesnik, M.A.; Atillasoy, E.; Thung, S.N.; Gerber, M.A. Identification of bipotential progenitor cells in human liver regeneration. Lab. Investig. 1996, 75, 699–705. [Google Scholar] [PubMed]
- Haruna, Y.; Saito, K.; Spaulding, S.; Nalesnik, M.A.; Gerber, M.A. Identification of bipotential progenitor cells in human liver development. Hepatology 1996, 23, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S. Hepatic stem cells in liver regeneration. FASEB J. 1996, 10, 1249–1256. [Google Scholar] [CrossRef]
- Sadri, A.R.; Jeschke, M.G.; Amini-Nik, S. Advances in Liver Regeneration: Revisiting Hepatic Stem/Progenitor Cells and Their Origin. Stem Cells Int. 2016, 2016, 7920897. [Google Scholar] [CrossRef] [Green Version]
- Nirmala, V.; Chopra, P.; Machado, N.O. An unusual adult hepatic teratoma. Histopathology 2003, 43, 306–308. [Google Scholar] [CrossRef]
- Verma, M.; Agarwal, S.; Mohta, A. Primary mixed germ cell tumour of the liver--a case report. Indian J. Pathol. Microbiol. 2003, 46, 658–659. [Google Scholar]
- Winter, T.C., 3rd; Freeny, P. Hepatic teratoma in an adult. Case report with a review of the literature. J. Clin. Gastroenterol. 1993, 17, 308–310. [Google Scholar] [CrossRef]
- Meyers, R.L. Tumors of the liver in children. Surg. Oncol. 2007, 16, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Moll, A.; Krenauer, A.; Bierbach, U.; Till, H.; Hirsch, W.; Leuschner, I.; Schmitz, N.; Wittekind, C.; Aigner, T. Mixed hepatoblastoma and teratoma of the liver in a 3-year-old child: A unique combination and clinical challenge. Diagn. Pathol. 2009, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.L.; Filippi, R.Z.; Zurakowski, D.; Archibald, T.; Vargas, S.O.; Voss, S.D.; Shamberger, R.C.; Davies, K.; Kozakewich, H.; Perez-Atayde, A.R. Effects of neoadjuvant chemotherapy on hepatoblastoma: A morphologic and immunohistochemical study. Am. J. Surg. Pathol. 2010, 34, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Hereditary tyrosinemia. General discussion: Screening aspects. Can. Med. Assoc. J. 1967, 97, 1096–1098.
- Forouhar, F.A.; Quinn, J.J.; Cooke, R.; Foster, J.H. The effect of chemotherapy on hepatoblastoma. Arch. Pathol. Lab. Med. 1984, 108, 311–314. [Google Scholar]
- Hiyama, E. Pediatric hepatoblastoma: Diagnosis and treatment. Transl. Pediatr. 2014, 3, 293–299. [Google Scholar] [CrossRef]
- Sergi, C.; Dhiman, A.; Gray, J.A. Fine Needle Aspiration Cytology for Neck Masses in Childhood. An Illustrative Approach. Diagnostics 2018, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, S.; Gupta, A.; Bansal, D.; Vora, T.; Prasad, M.; Arora, B.; Kapoor, G.; Chinnaswamy, G.; Radhakrishnan, V.; Laskar, S.; et al. Management of Hepatoblastoma: ICMR Consensus Document. Indian J. Pediatr. 2017, 84, 456–464. [Google Scholar] [CrossRef]
- Aronson, D.C.; Czauderna, P.; Maibach, R.; Perilongo, G.; Morland, B. The treatment of hepatoblastoma: Its evolution and the current status as per the SIOPEL trials. J. Indian Assoc. Pediatr. Surg. 2014, 19, 201–207. [Google Scholar] [CrossRef]
- Pimpalwar, A.P.; Sharif, K.; Ramani, P.; Stevens, M.; Grundy, R.; Morland, B.; Lloyd, C.; Kelly, D.A.; Buckles, J.A.; de Ville De Goyet, J. Strategy for hepatoblastoma management: Transplant versus nontransplant surgery. J. Pediatr. Surg. 2002, 37, 240–245. [Google Scholar] [CrossRef]
- Ismail, H.; Broniszczak, D.; Kalicinski, P.; Dembowska-Baginska, B.; Perek, D.; Teisseyre, J.; Kluge, P.; Kosciesza, A.; Lembas, A.; Markiewicz, M. Changing treatment and outcome of children with hepatoblastoma: Analysis of a single center experience over the last 20 years. J. Pediatr. Surg. 2012, 47, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Hino, M.; Koh, K.; Kyushiki, M.; Kishimoto, H.; Arakawa, Y.; Hanada, R.; Kawashima, H.; Kurihara, J.; Shimojo, N.; et al. Low Frequency of Programmed Death Ligand 1 Expression in Pediatric Cancers. Pediatr. Blood Cancer 2016, 63, 1461–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergi, C.M.; Rojas-Vasquez, M.; Noga, M.; Dicken, B. ‘Teratoid’ Hepatoblastoma: An Intriguing Variant of Mixed Epithelial-Mesenchymal Hepatoblastoma. Children 2022, 9, 565. https://doi.org/10.3390/children9040565
Sergi CM, Rojas-Vasquez M, Noga M, Dicken B. ‘Teratoid’ Hepatoblastoma: An Intriguing Variant of Mixed Epithelial-Mesenchymal Hepatoblastoma. Children. 2022; 9(4):565. https://doi.org/10.3390/children9040565
Chicago/Turabian StyleSergi, Consolato M., Marta Rojas-Vasquez, Michelle Noga, and Bryan Dicken. 2022. "‘Teratoid’ Hepatoblastoma: An Intriguing Variant of Mixed Epithelial-Mesenchymal Hepatoblastoma" Children 9, no. 4: 565. https://doi.org/10.3390/children9040565
APA StyleSergi, C. M., Rojas-Vasquez, M., Noga, M., & Dicken, B. (2022). ‘Teratoid’ Hepatoblastoma: An Intriguing Variant of Mixed Epithelial-Mesenchymal Hepatoblastoma. Children, 9(4), 565. https://doi.org/10.3390/children9040565