2.1. Modeling and Calculation Methods
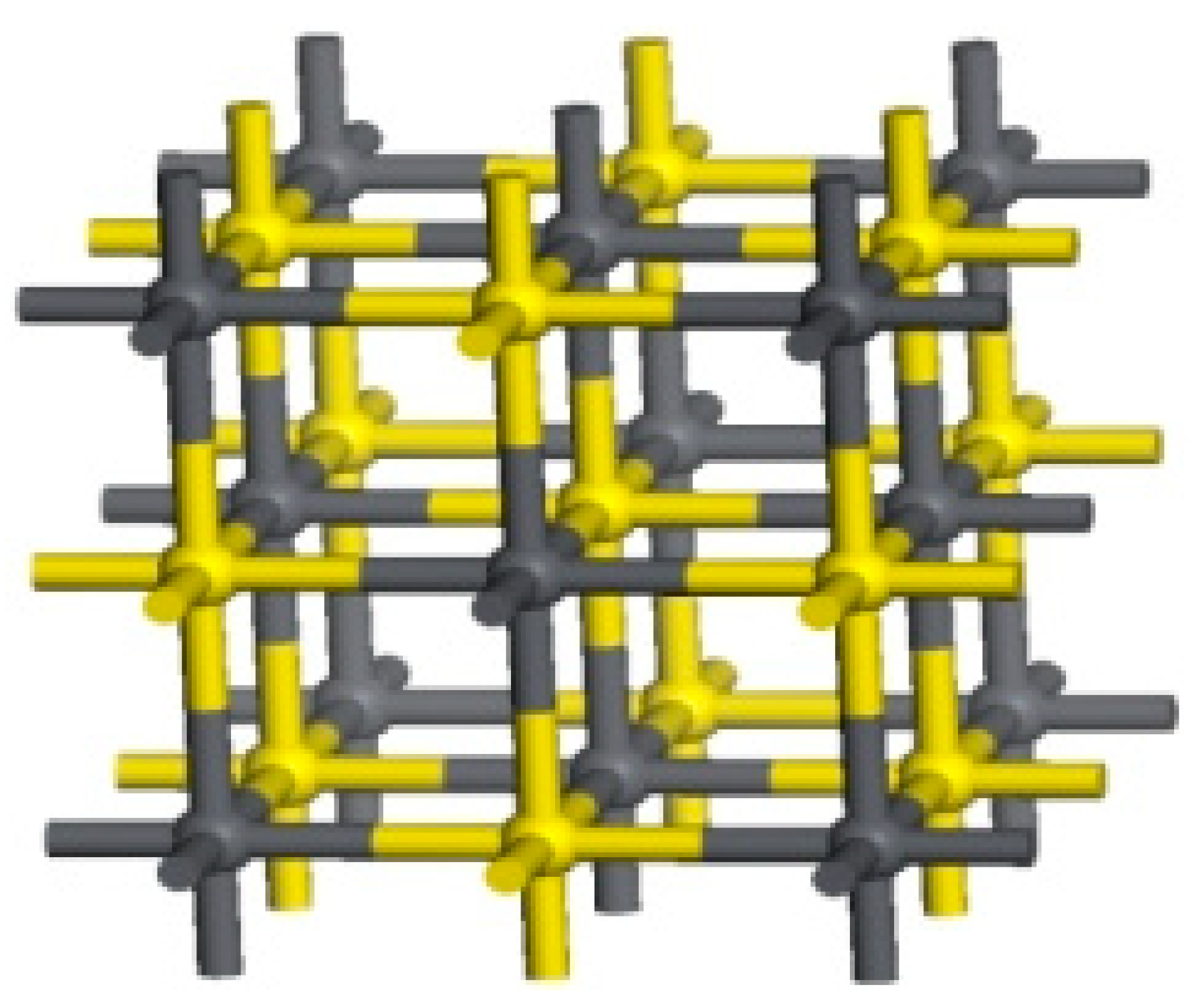
Galena belongs to the cubic crystal system; its crystal is a NaCl-type and belongs to the face-centered cubic lattice. Galena has a symmetrical type of (3L44L36L29PC) and displays hexagonal symmetry. In the galena lattice, the S atoms are the closest cubic packing, and the Pb atoms fill in the octahedral gap with a coordination number of 6. The (100) plane of the crystal can be most wholly dissociated, which belongs to the space group: O5H-Fm3m; the lattice parameters: a= b = c =5.9315 Å, α = β = γ = 90°.
The ideal galena lattice model constructed according to known parameters is shown in
Figure 1.
To make the lattice constant and the band gap of the model closer to the experimental value, it is necessary to optimize the original cell of galena and finally determine the best exchange-correlation functions and energy cut-off. In this study, the CASTEP module based on density functional in the Materials Studio software was employed for calculation. For the optimization process of the galena lattice system, researchers mainly adopt the following geometric optimization criteria: exchange-correlation functions and energy cut-off were GGA-PW91 and 300 eV, respectively. In addition, the convergence criteria of geometric optimization were set as follows: atomic displacement was 0.002 Å, interatomic force was 0.05 eV·Å, interatomic internal stress was 0.1 GPa, maximum energy change was 2.0 × 10
−5 eV/atom; self-consistent iteration was 2.0 × 10
−6 eV/atom; convergence accuracy of self-consistent iteration was 2.0 × 10
−6 eV/atom; smearing value calculated by state density property was 0.1 eV. The valence electrons selected for the pseudopotential calculation of atoms are 5d
106s
26p
2 of Pb and 3s
23p
4 of S [
27,
28].
The (100) surface of the galena was cut out from the optimized galena cell. A vacuum layer of 10 Å was added to eliminate the influence of periodic boundaries. The CASTEP module in the Materials Studio software was adopted to test the convergence of atomic layer numbers on galena surfaces based on first-principles density functional theory, which obtained a surface model consistent with practice. The exchange-correlation functions and the plane wave cut-off energy were selected from the convergence results of galena crystal cells.
The number of atomic layers was determined by studying the surface energy of galena with different atomic layers. When the number of atomic layers is different, the surface energy of the galena surface is calculated according to the following formula [
29]:
is the unit area of the surface energy, represents the total free energy of the slab, n is the total number of atoms in the slab, is the free energy of each atom inside the crystal, and A represents the unit area of the surface.
2.2. Crystal Structure Analysis of Galena
The lattice protocell model of galena is optimized by changing different plane wave cut-off energies and using existing optimization parameters. The cut-off energy with the minimum final energy of the cell is selected to determine the optimal cut-off energy of the plane wave in the optimization process. The software simulation results are shown in
Figure 2.
As shown in
Figure 2, the lattice constant and final energy of the cell decrease with the increase in the plane wave cut-off energy, reaching the lowest level when the cut-off energy was 300 eV. At this point, the lattice constant of 5.999 Å is closest to the experimental value of 5.9315 Å. The larger the plane wave cut-off energy is, the more accurate the lattice constants will be, but the amount of software calculation is also increasing. Therefore, the cut-off energy is selected as 300 eV.
The plane wave cut-off energy of 300 eV is selected, and the remaining parameters are unchanged to obtain the best exchange-correlation function. By changing different exchange-correlation functions and using software for simulation calculation, the best exchange-correlation function was determined in the optimization process of galena crystal. The simulation results are presented in
Table 1.
As is evident from
Table 1, the band gap of the exchange-correlation functions LDA-CA-PZ, GGA-RPBE, and GGA-WC groups have a significant large deviation from the experimental value of 0.41 eV. Although the final energy of the cell of the exchange-correlation function GGA-PBE is lower than GGA-PW91, the lattice constant and band gap width of GGA-PW91 are closer to the standard value. Therefore, the exchange-correlation function GGA-PW91 is selected comprehensively.
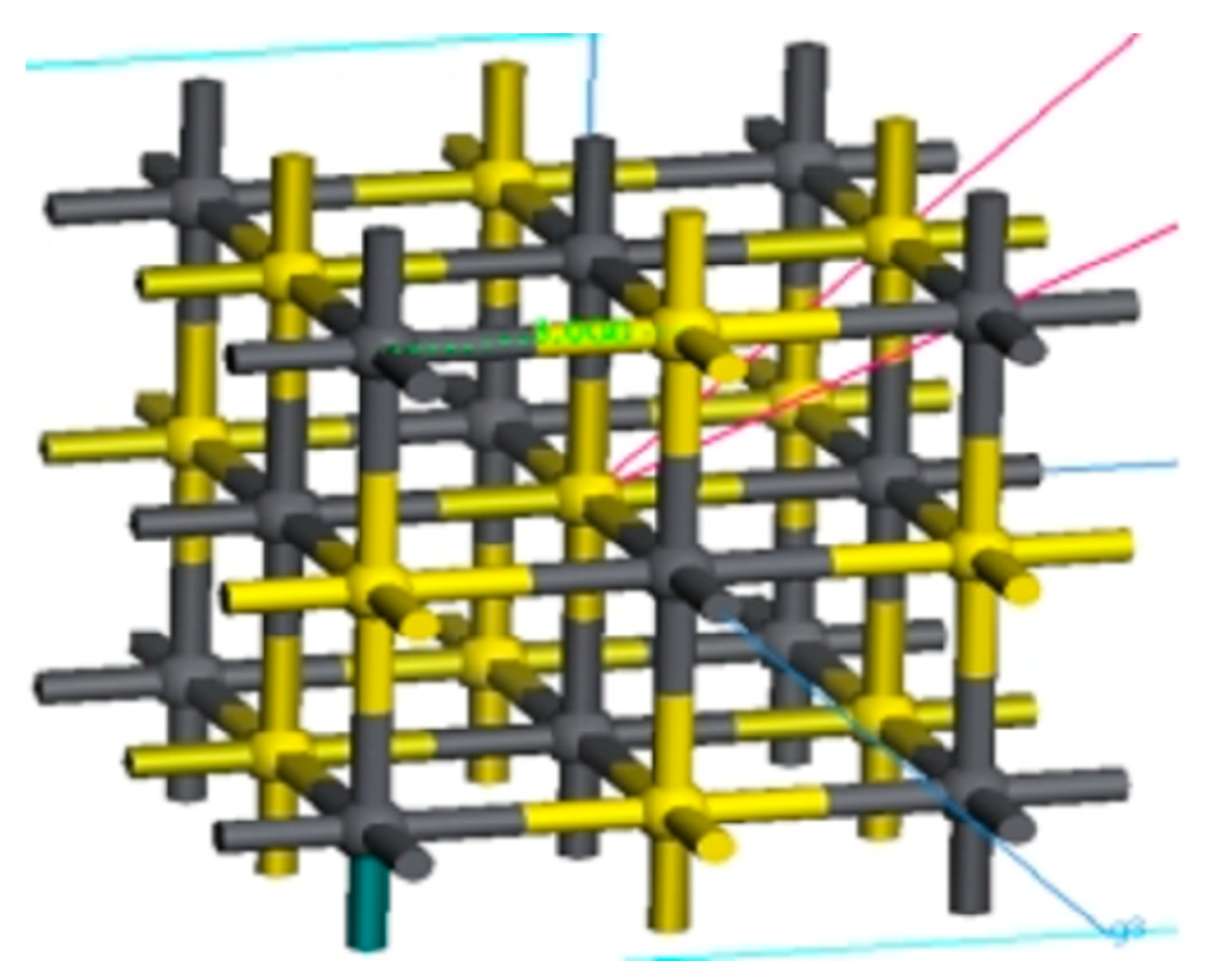
In the crystal structure of galena, the bond distance of Pb-S was 2.966 Å obtained from known lattice parameters, and the structure optimization was carried out. With the rest remaining unchanged, the exchange-correlation function and energy cut-off in the geometric optimization standard are GGA-PW91 and 300 eV, respectively. The bond distance of Pb-S in the crystal structure of galena after structural optimization was 3.000 Å as shown in
Figure 3.
Table 2 compares the theoretical and experimental values of the lattice constant and band gap of galena under ideal conditions. It can be seen that the experimental lattice constant value of galena crystal is 0.5999 nm, which is very close to the theoretical value of 0.5931 nm. This shows that the model selected for the simulation and optimization of the galena lattice model is reasonable.
Chen et al. [
30], using the Generalized gradient approximation (GGA) method to simulate the calculation of electronic structure properties of galena, examined the different exchange-correlation function effects on its lattice parameters (
Table 3). The lattice parameters of galena: a = b = c = 5.9324 Å, α = β = γ = 90°.
As is evident from
Table 1 and
Table 3, the band gaps of the exchange-correlation functions GGA-RPBE and GGA-PBE both have a significantly large deviation from the experimental value of 0.41 eV. From the exchange functions of GGA-PW91 and GGA-WC, it is obvious that the results in this paper are more accurate than those of Chen et al. Therefore, our calculations are without spin-orbit coupling (SOC).
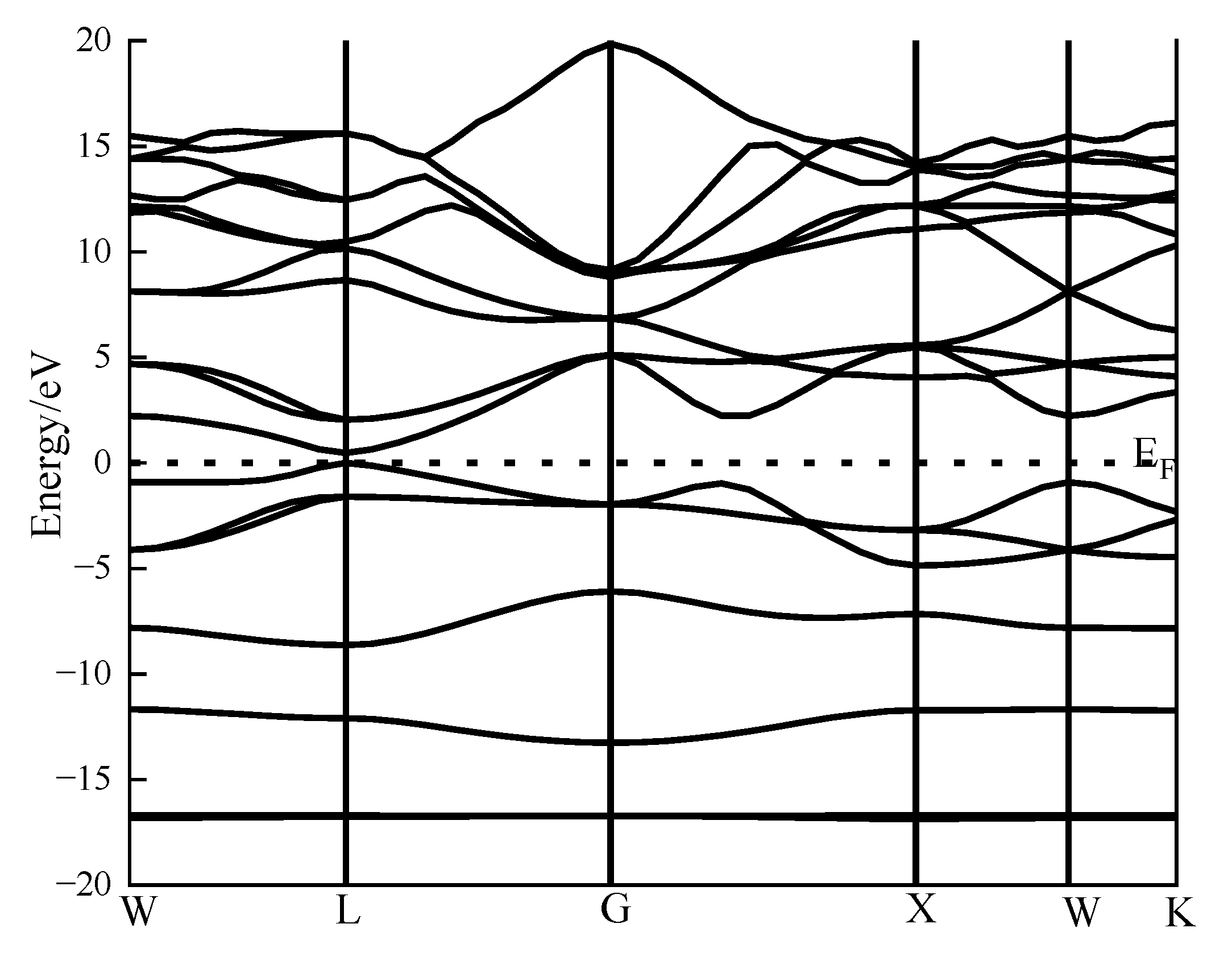
Figure 4 and
Figure 5 show the band structure and density of states of perfect galena, calculated using CASTEP module simulation.
As demonstrated in
Figure 4, the maximum value of the valence band and the minimum value of the conduction band in the band structure diagram of galena are both located at the highly symmetric G point, confirming that galena is a direct band-gap P-type semiconductor.
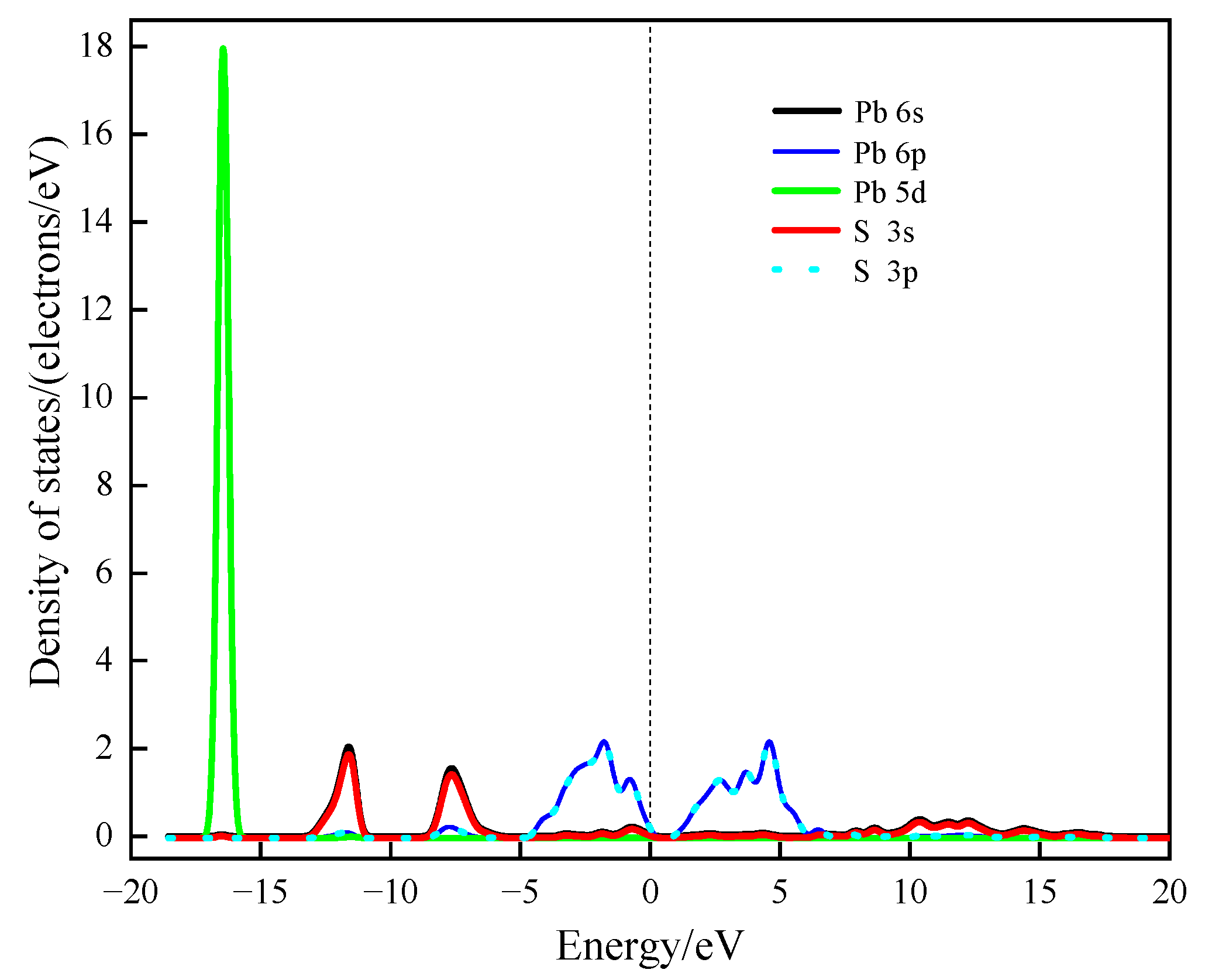
According to the density of states distribution diagram of galena in
Figure 5, the upper valence band and the guide band are mainly composed of the 5d orbital of the Pb atom and the 3p orbital of the S atom. The lower valence band is comprised of the 5d orbital of the Pb atom.
Mulliken population distribution reflects the overlap of electrons between two atoms, providing a criterion for ionic and covalent bonding between them. The value of the Mulliken overlapping population indicates the degree of charge overlap between atoms, that is, the covalent strength of bonds between atoms. The results of the Mulliken population analysis of galena atoms are shown in
Table 4.
As can be seen from
Table 4, the valence electron configuration of the S atom is 3s
23p
4 before structural optimization, while the optimized valence electron configuration of the S atom is 3s
1.923p
4.73. The number of electrons localized in the S atom is 6.64 e, so the S atom obtains 0.64 e. Because its charge is −0.64e, it is an electron acceptor. It can be seen that the electrons are mainly obtained from the 3p orbital of the S. Before the structural optimization, the valence electron configuration of the Pb atom is Pb 5d
10 6s
2 6p
2. In contrast, the optimized valence electron configuration of the Pb atom is Pb 5d
106s
3.926p
1.44. The number of electrons localized in the Pb atom is 13.36, so the Pb atom decreases by 0.64e. Because its charge is +0.64 e, it is the electron donor. It is mostly losing electrons in the 6s and 6p orbitals of Pb. In addition, the 3p orbital of S and the 6p orbital of Pb contribute to the formation of PbS crystals from the data in the table.
Table 5 shows the atomic orbital coefficients composed of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of galena electrons. The positive and negative signs of the coefficients represent the bonding and antibonding between atoms. The absolute values of the coefficients indicate the contribution to frontier orbits. Ordinarily, the most easily transferred orbit of electrons is the highest orbit occupied by electrons, while the most easily obtained orbit is the lowest unoccupied one. In short, the HOMO and LUMO orbitals are nucleophilic and electrophilic, respectively. According to the absolute value of the atomic coefficient in
Table 5, it can be found that S atoms mainly contribute to the highest orbital. In contrast, Pb atoms contribute primarily to the lowest unoccupied orbital. Therefore, S atoms in galena are easy to lose electrons and undergo oxidation, while Pb atoms are easy to interact with anions.
2.3. Surface Structure and Electronic Properties of Galena
In this study, the relationship between atomic layers and surface energy was investigated using the surface of galena (100) of 431 super crystal cells. The results are indicated in
Figure 6.
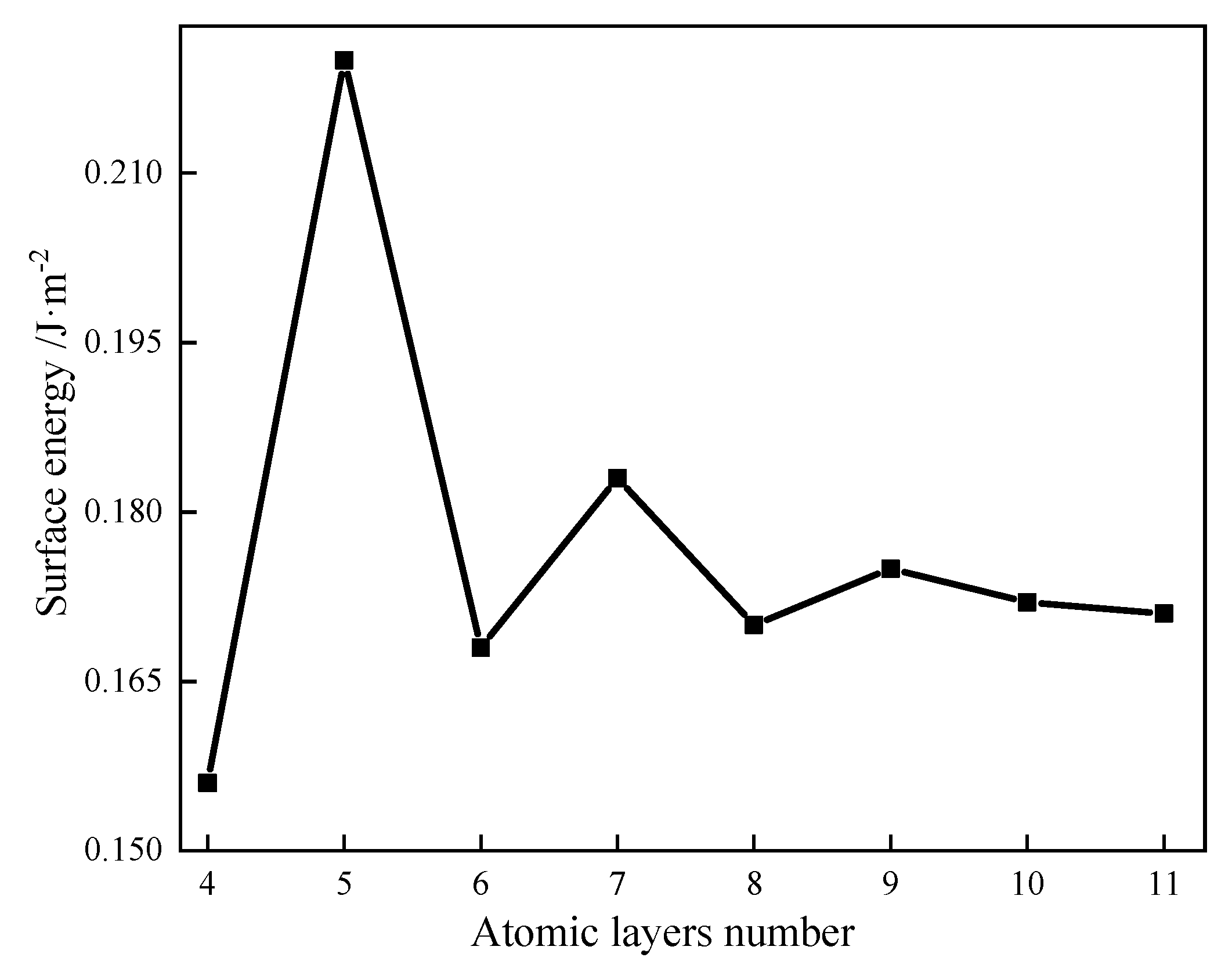
As demonstrated in
Figure 6, when the atomic layer number is small, the variation range of the surface energy of galena decreases gradually with the increase in the atomic layer number. When the atomic layer number is 8, the surface energy change tends to be gentle. Therefore, the atomic layer number and thickness of the vacuum layer of the galena surface model are 8 and 15 Å, respectively.
The solid crystal structure is a stable periodic lattice structure. The mineral surface is exposed by breaking part of the connecting bonds, resulting in a structure different from the internal atomic environment. The force on such an aperiodic surface structure is no longer symmetrical. To achieve a new equilibrium, the phenomenon of readjusting the spacing of surface atoms is called surface relaxation [
31].
The dissociation of the galena surface causes the atomic coordination number of Pb and S to decrease to five, which weakens the binding force of galena surface atoms, resulting in surface relaxation.
Table 6 shows the displacement of the three layers of atoms on the galena surface before and after optimization.
As indicated in
Table 6, the relaxation of Pb and S atoms in the three layers on the surface mainly occurs in the vertical direction, while the relaxation displacement in the horizontal direction is small. In general, the relaxation of the ideal (100) surface of the galena is minimal. There is no evident surface reconstruction, so during the surface optimization calculation, only the lower five layers of atoms need to be fixed to relax the three layers of atoms on the surface. At this time, the calculation can be reduced under the condition of ensuring accuracy.
Figure 7 shows the crystal plane model before and after surface relaxation on the (100) cleavage surface of the galena.
The environment of the atoms on the surface layer is different from that in the bulk, which is unsaturated and shows vigorous surface-reaction activity. The band gap is an essential characteristic parameter of semiconductors, and its size is related to the band structure of semiconductors. In other words, the band gap is associated with the crystal structure and atomic binding energy. The band structure of bulk galena (a) and (100) surface (b) are displayed in
Figure 8.
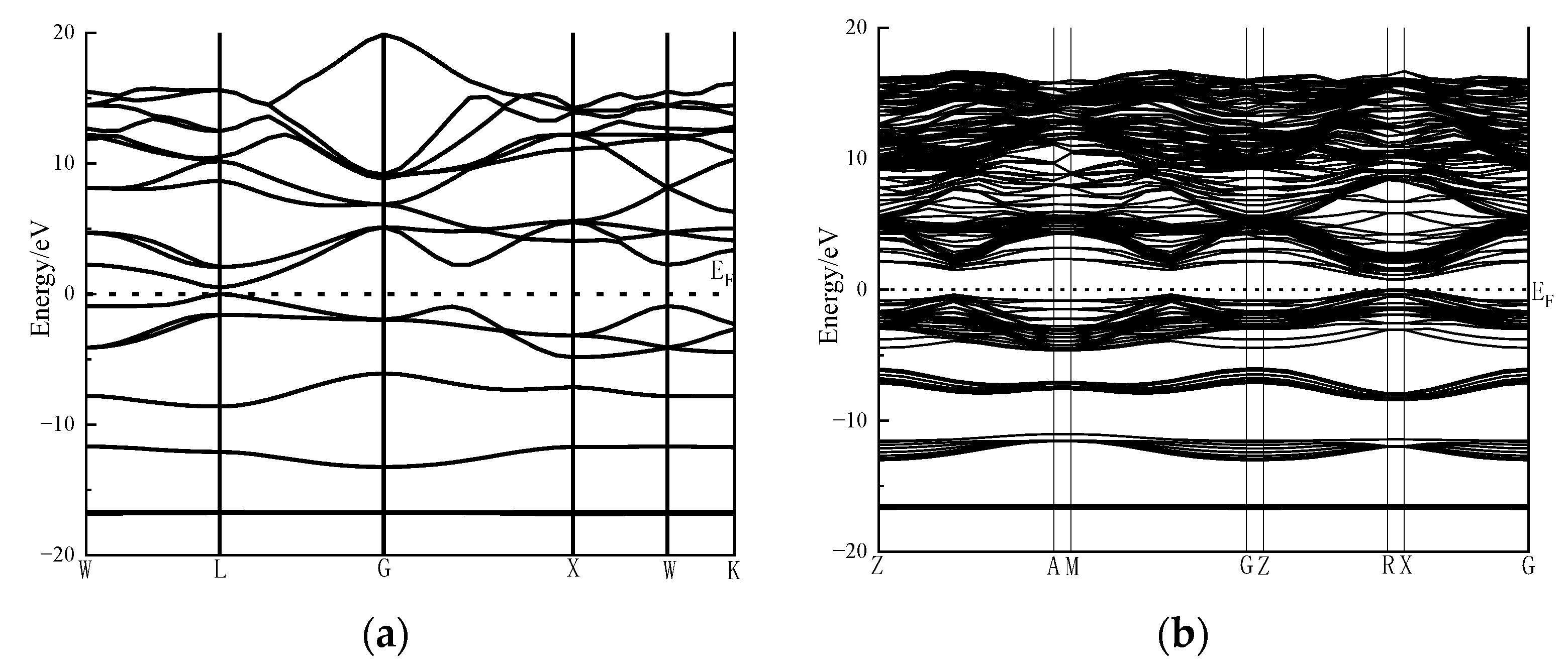
As presented in
Figure 8, when more atoms are used, the energy band curves of the galena surface model are denser than those of the bulk model. The simulation results show that the band gap of the galena bulk and the surface is 0.478 eV and 0.787 eV, respectively. The change in the band gap indicates that the electronic structure of the galena surface has changed significantly. Meanwhile, the electron transition from the valence band to the conduction band requires more energy.
According to previous research on the relaxation of galena surface structure, it was found that the galena surface undergoes significant relaxation. Therefore, it can be inferred that although the coordination number of atoms on the surface of galena is reduced compared to the bulk, the change of coordination number is weakened due to the large atomic number of Pb atoms and strong binding effect on valence electrons. The immense relaxation on the surface structure of galena has a marked impact on the binding of valence electrons, leading to an increase in electron transition energy. Therefore, the electronic activity of the galena surface is weakened.
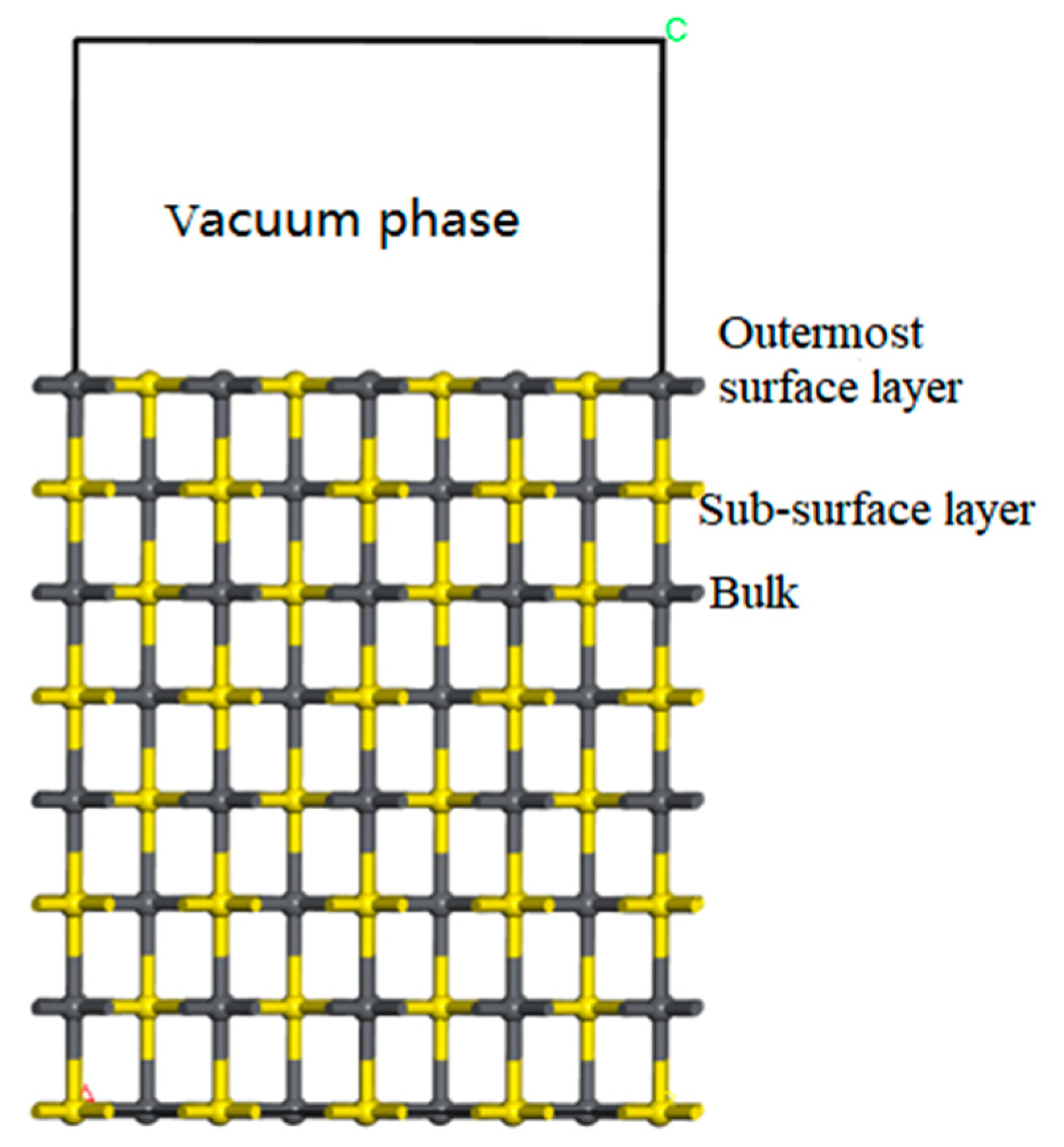
Figure 9 shows the outermost surface layer, sub-surface layer, and bulk on the surface (100) of the galena.
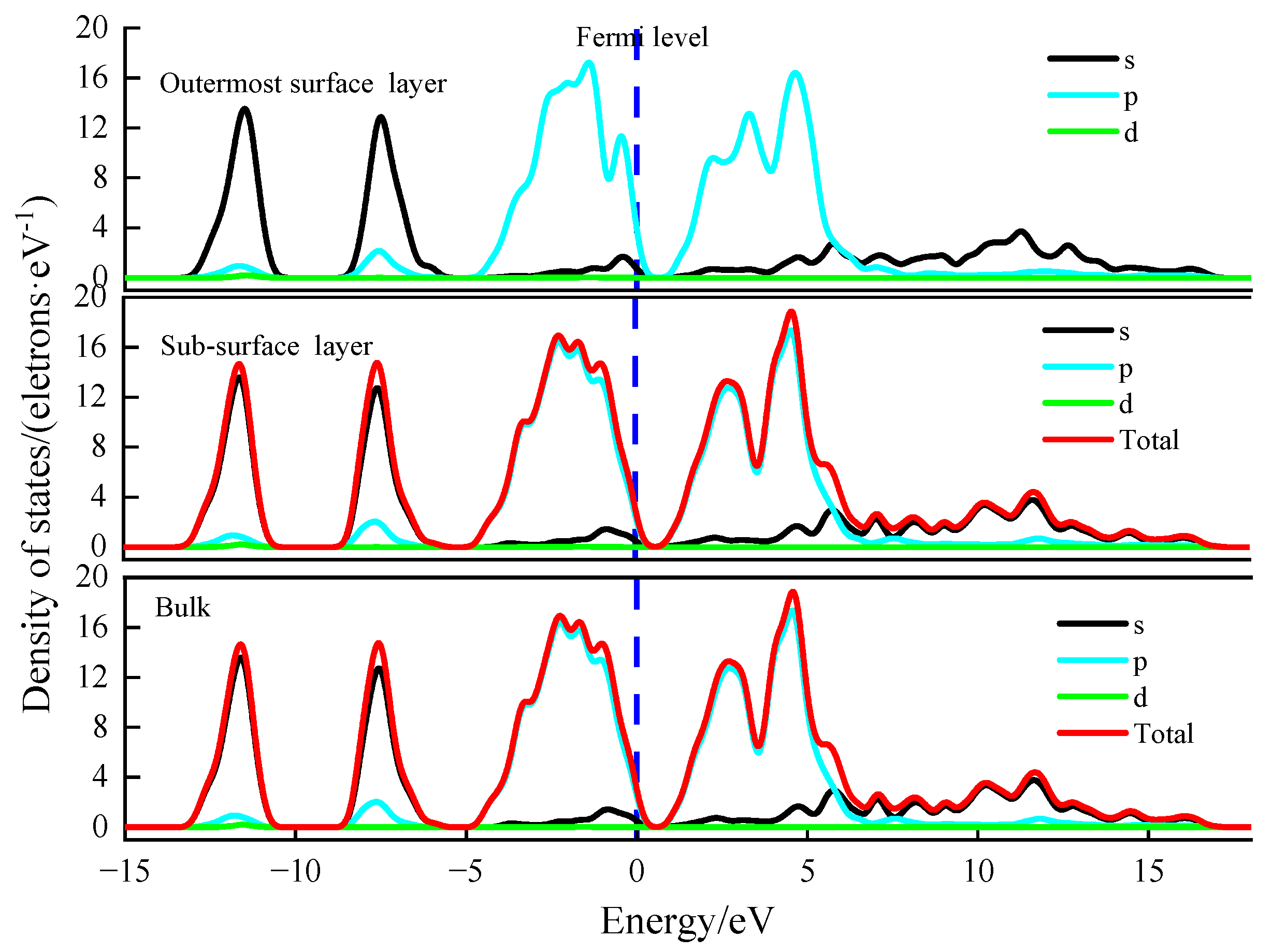
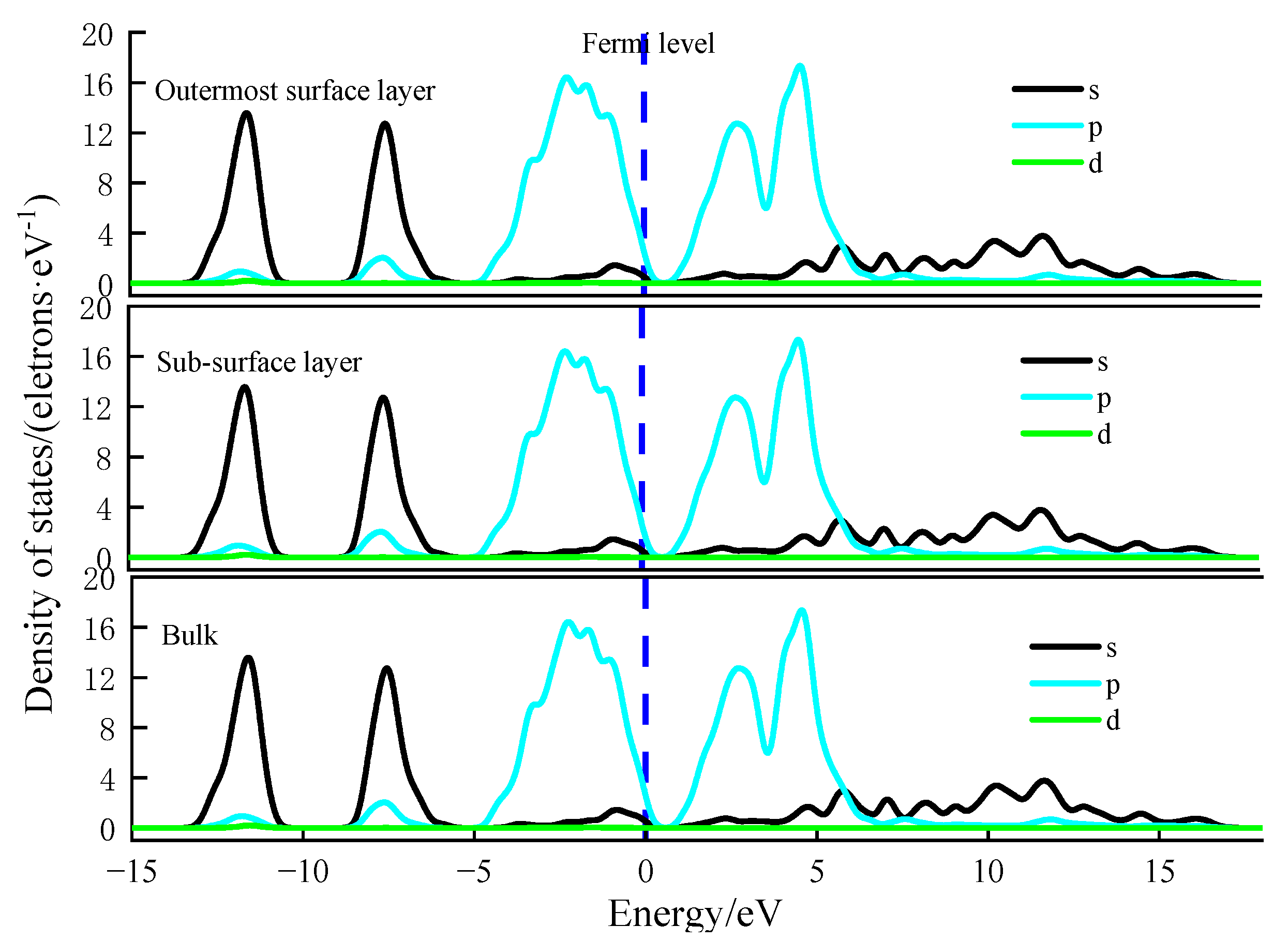
Figure 10 and
Figure 11 are acquired by calculating the state density with Pb atoms and S atoms in the outermost surface layer, sub-surface layer, and bulk.
As can be seen in
Figure 10, the density of S atomic states in the outermost surface layer, sub-surface layer, and bulk on the (100) surface of galena are basically similar, which is consistent with the atomic relaxation results on galena surface.
It can be seen from the density of states of Pb atoms of the outermost surface layer, sub-surface layer, and bulk on (100) surface of galena in
Figure 11 that the 6p density of states peak of the Pb and S atoms in these layers is the strongest at −2 eV, which shows that the Fermi level is mainly affected by the 6p orbital of Pb.
The positions of Pb and S atoms in each layer of the galena lattice are at the same height. In addition, the structure of their surface is relatively uncomplicated.
Table 7 shows the electronic changes of the three layers of the galena surface after surface relaxation.
As presented in
Table 7, the charge of the S atom changes from −0.71 e in the bulk to −0.61e in the outermost surface layer. In comparison, the charge of the Pb atom decreases from 0.7e in the bulk to 0.67 e in the sub-surface layer and then to 0.61 e in the outermost surface layer. From the perspective of electron orbital distribution, the valence electron structure adopted in the calculation is 5d
106s
26p
2 for the Pb atom and 3s
23p
4 for the S atom. The analysis found that the 3p orbital of S and the 6p orbital of Pb mainly play a role in galena crystal. Ordinarily, electrons from the 6p orbital of Pb will transfer to the 3p orbital of S, resulting in the change of atomic valence states on the lattice surface. It can also be seen from
Table 6 that, in the process of relaxation of the galena surface structure, electrons change from 4.74 in the bulk to 4.75 in the outermost surface layer in the 3p orbital of S, and electrons are both 1.41 in the bulk and outermost surface layer in 6p orbital of Pb. These indicate that the role of the 3p orbital of S and 6p orbital of Pb will not be affected in this process. However, the electrons in the 6s orbital of Pb in the outermost surface layer change from 6s
1.88 in the bulk to 6s
1.98 on the outermost surface layer surface. According to the electron distribution and charge change on the galena surface, the total electron number of the outermost surface layer was higher than that of the bulk, so the galena surface owns the properties of electron enrichment.


 ,
, 











{kind=link}
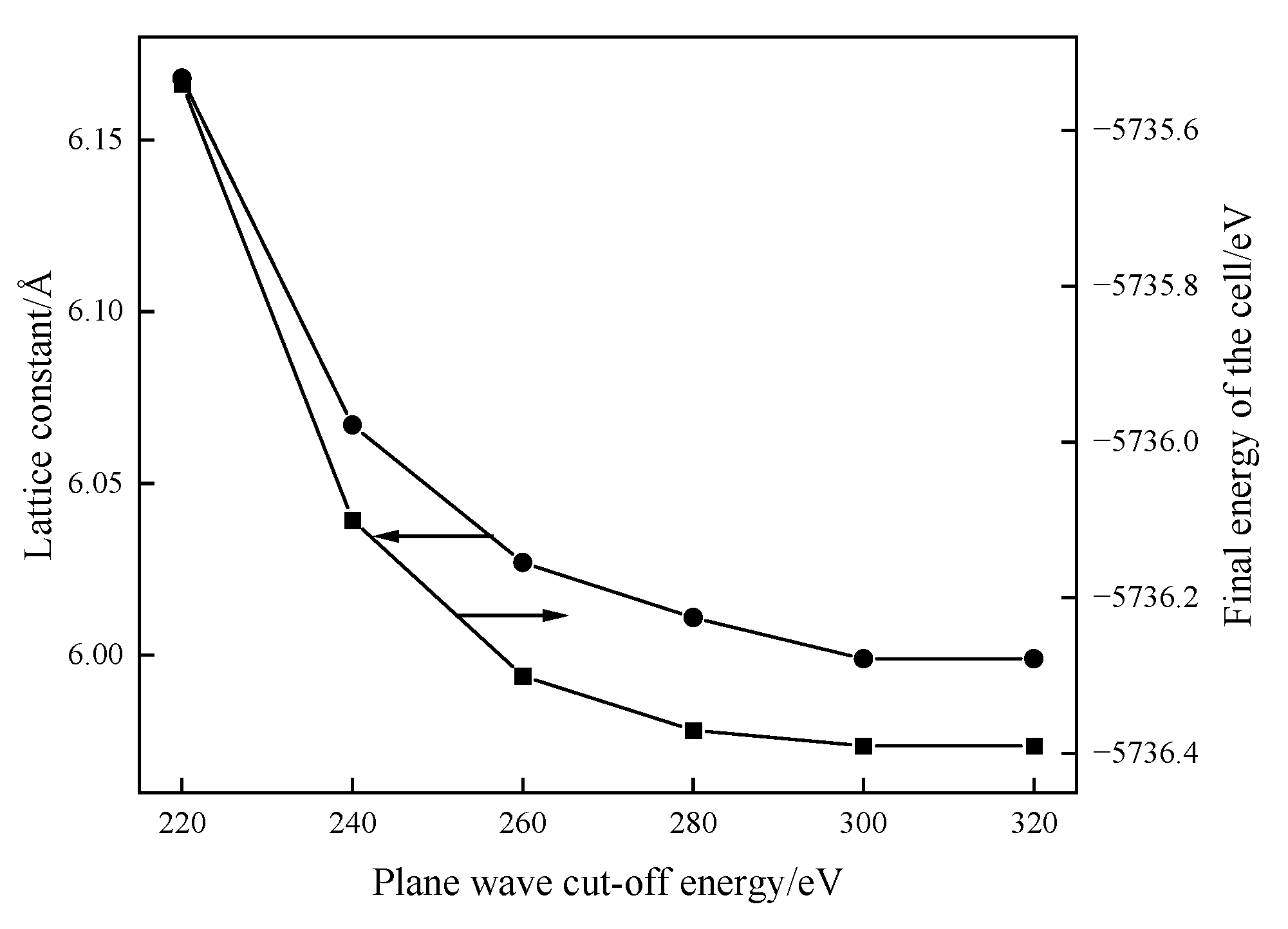{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}