
Simultaneous Monitoring of the Effects of Multiple Ionic Strengths on Properties of Copolymeric Polyelectrolytes during Their Synthesis


Abstract
:1. Background and Motivation
1.1. Background on Solution Properties of Polyelectrolytes in Solution
1.2. Background on ACOMP
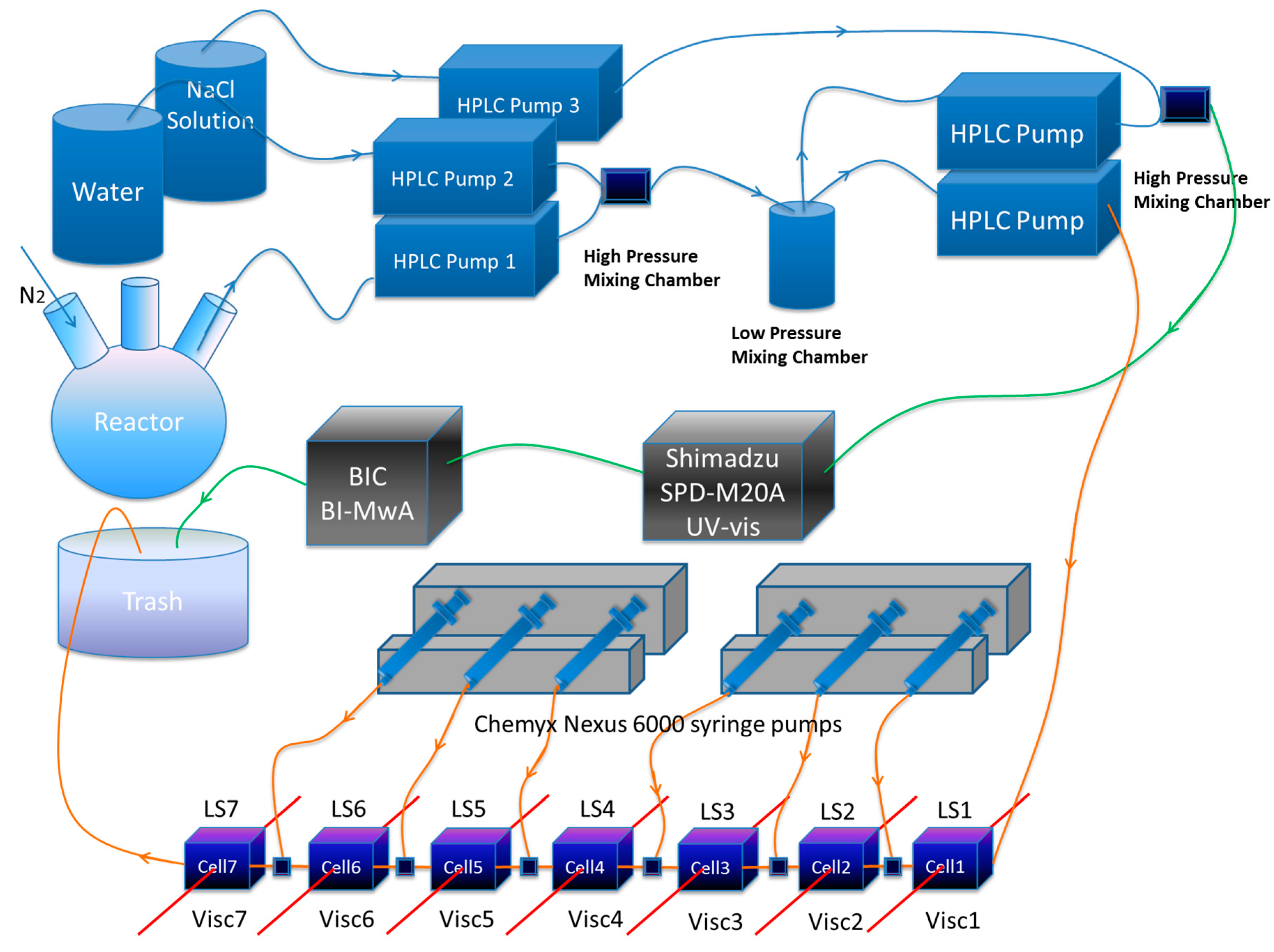
2. Second Generation ACOMP (SGA) for Monitoring the Synthesis of Stimuli Responsive Polymers under Varying Solution Conditions
3. Copolymerization
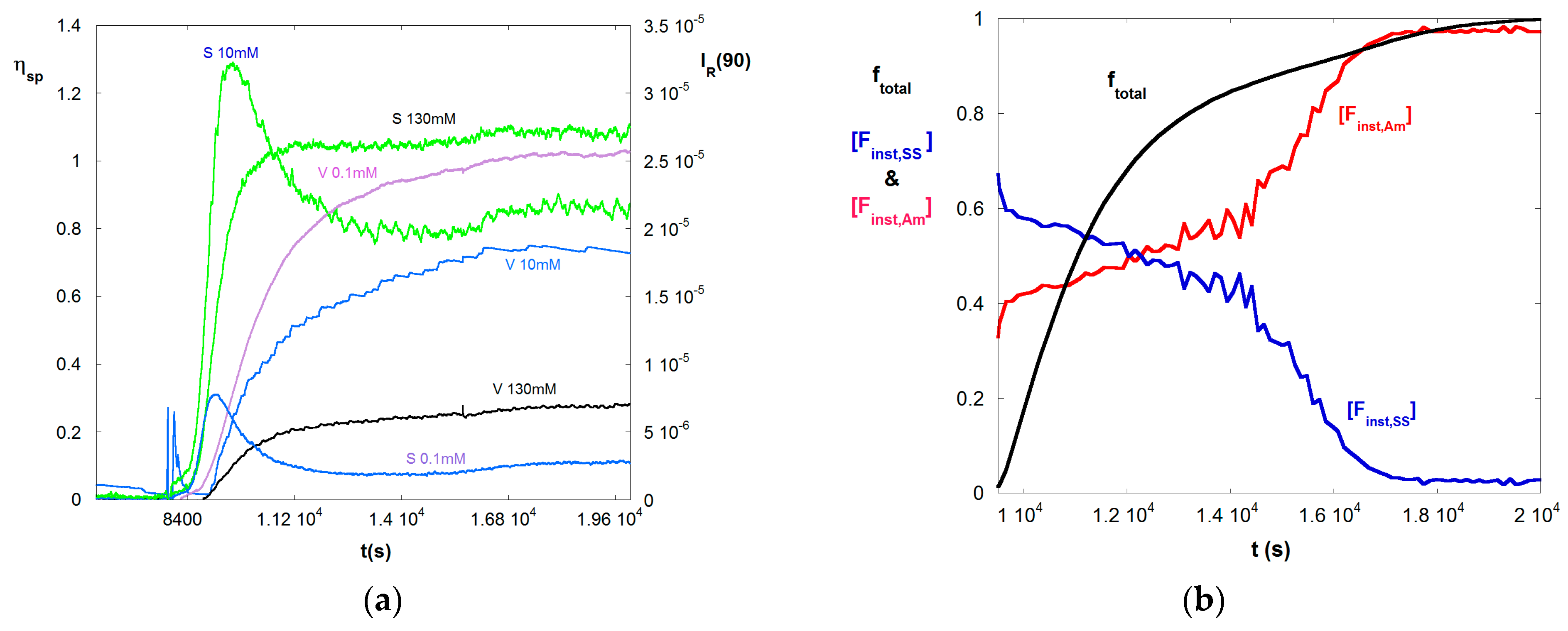
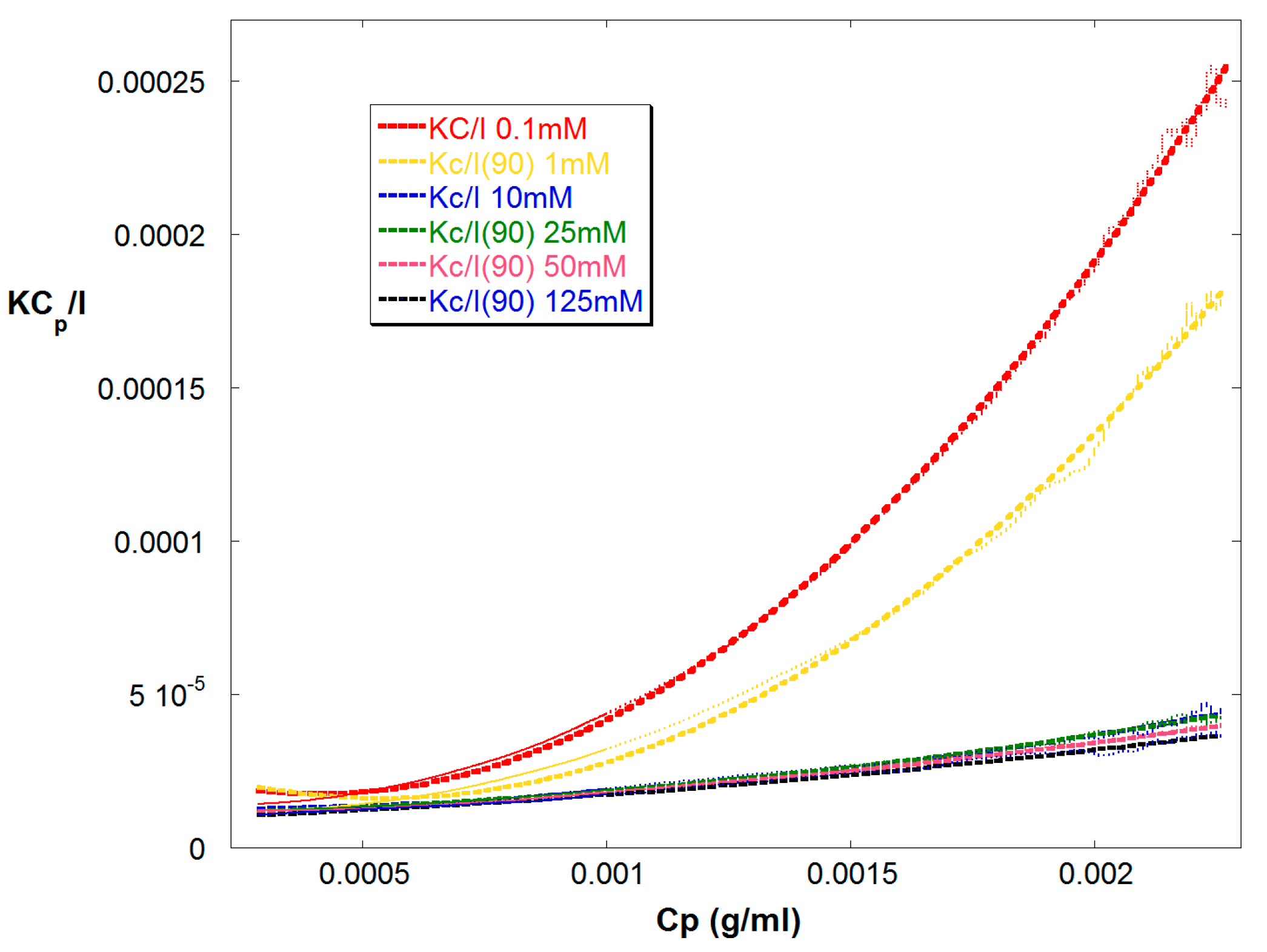
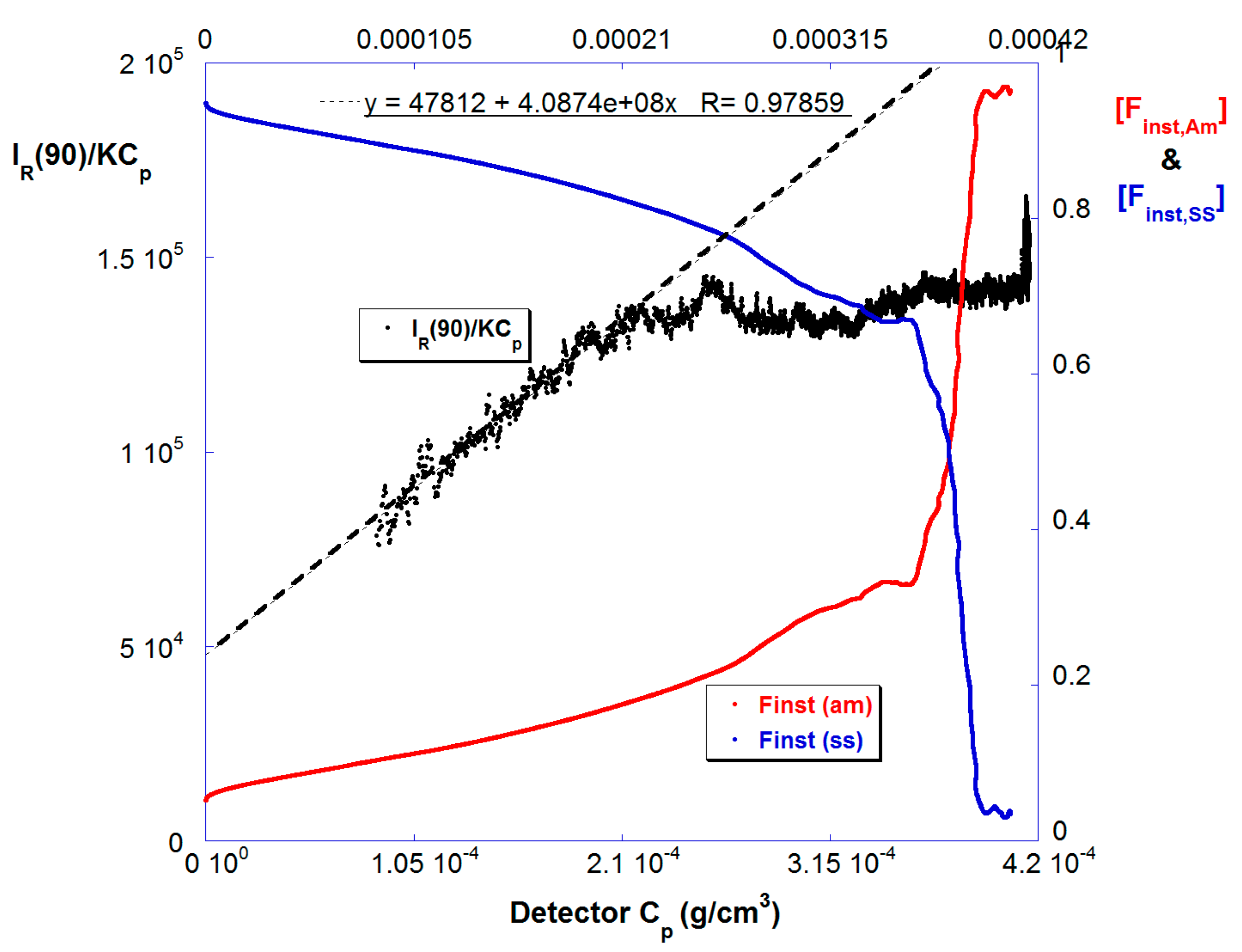
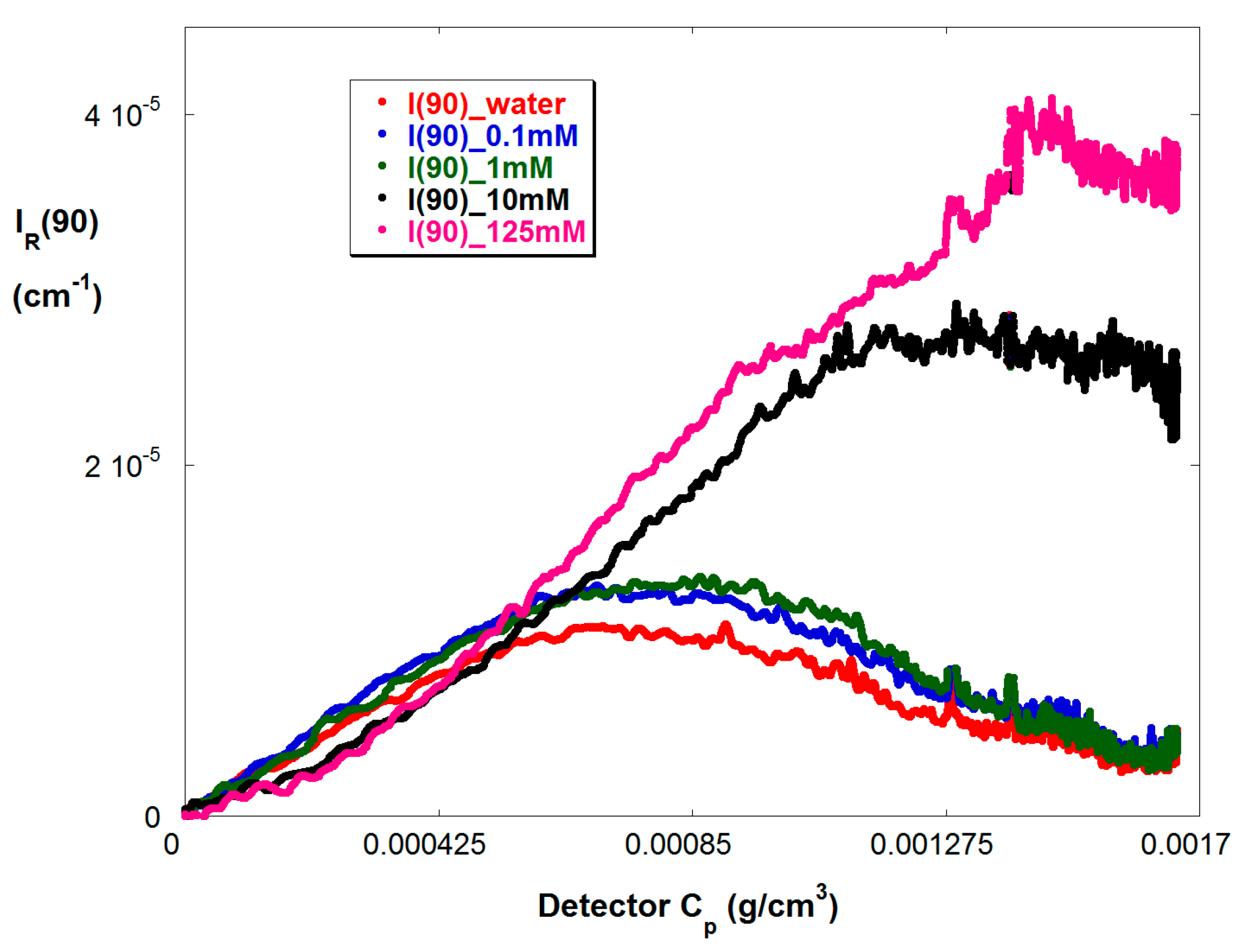
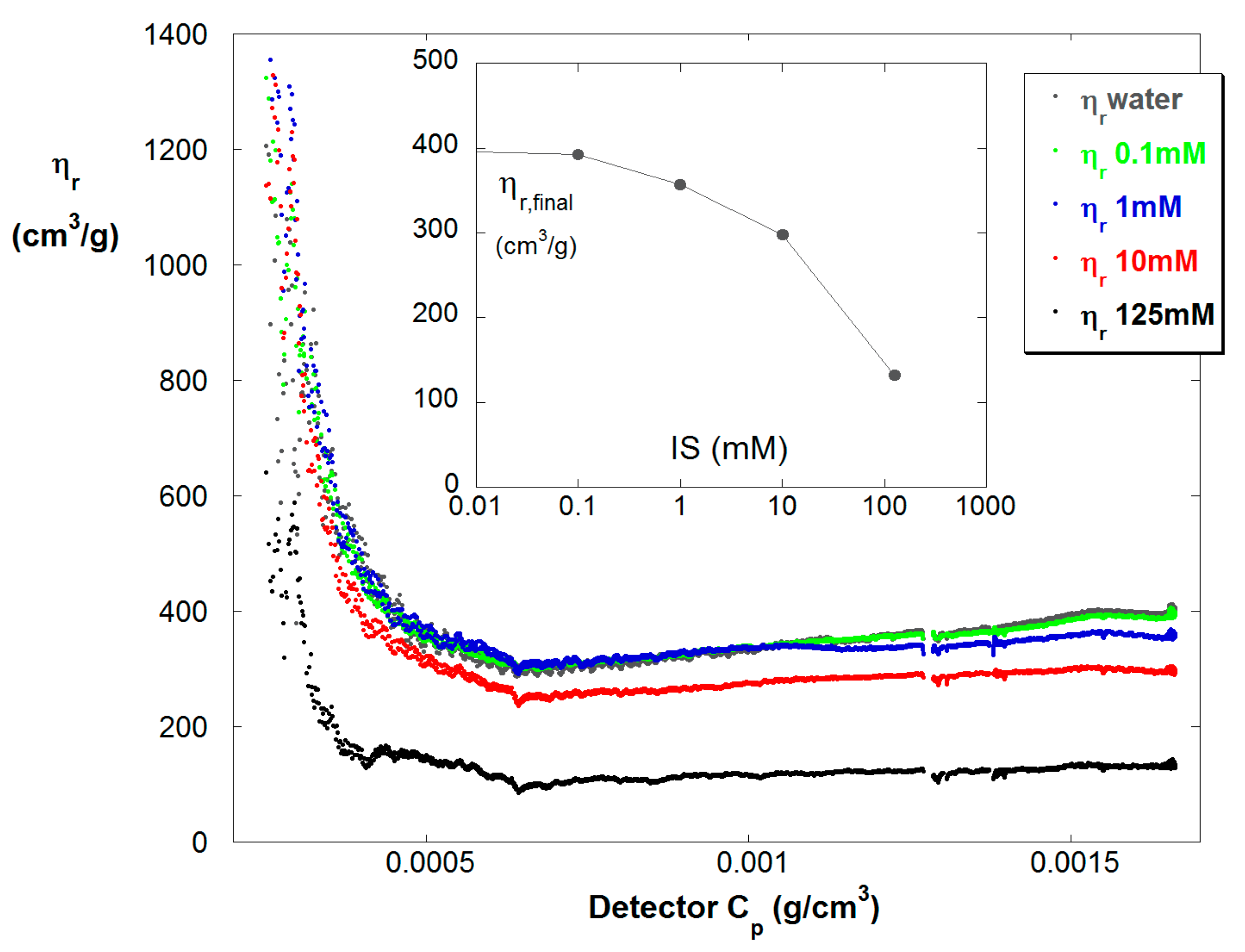
4. Results and Discussion
4.1. Typical Raw Data
4.2. SGA Data Analysis
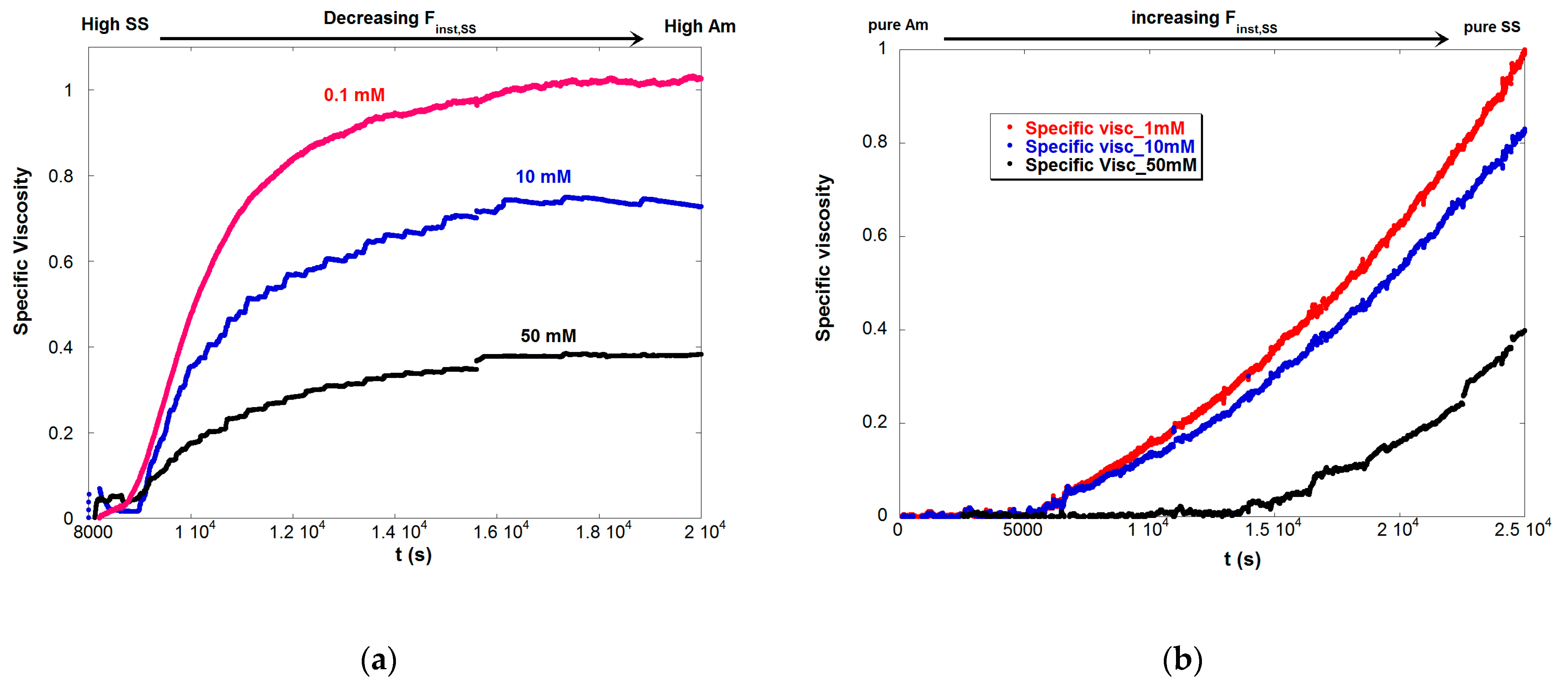
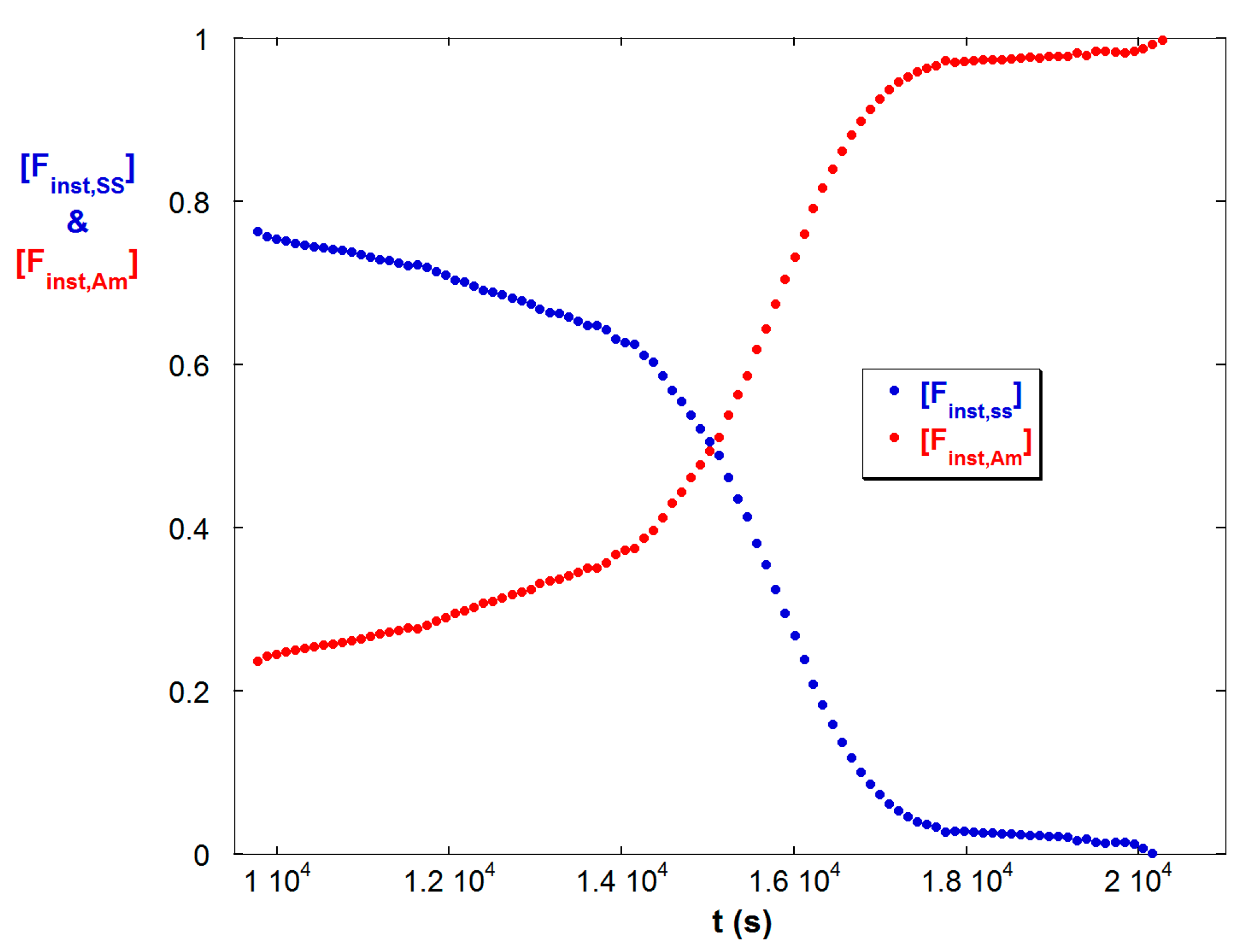
4.3. Contrasting Viscosity Behavior in a Low to High ξ Reaction with a High to Low ξ Reaction
5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhuang, J.; Gordon, M.R.; Ventura, J.; Li, L.; Thayumanavan, S. Multi-stimuli responsive macromolecules and their assemblies. Chem. Soc. Rev. 2013, 42, 7421–7435. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Gutowska, A. Lessons from nature: Stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002, 20, 305–311. [Google Scholar] [CrossRef]
- Nath, N.; Chilkoti, A. Creating “smart” surfaces using stimuli responsive polymers. Adv. Mater. 2002, 14, 1243–1247. [Google Scholar] [CrossRef]
- Liu, F.; Urban, M.W. Recent advances and challenges in designing stimuli-responsive polymers. Prog. Polym. Sci. 2010, 35, 3–23. [Google Scholar] [CrossRef]
- Galaev, I.; Mattiasson, B. Smart Polymers: Applications in Biotechnology and Biomedicine; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Chung, J.E.; Yokoyama, M.; Yamato, M.; Aoyagi, T.; Sakurai, Y.; Okano, T. Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly(n-isopropylacrylamide) and poly(butylmethacrylate). J. Control. Release 1999, 62, 115–127. [Google Scholar] [CrossRef]
- Zhang, J.; Peppas, N.A. Synthesis and characterization of pH-and temperature-sensitive poly(methacrylic acid)/poly(n-isopropylacrylamide) interpenetrating polymeric networks. Macromolecules 2000, 33, 102–107. [Google Scholar] [CrossRef]
- Jain, S.; Bates, F.S. On the origins of morphological complexity in block copolymer surfactants. Science 2003, 300, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Sumerlin, B.S.; Tsarevsky, N.V.; Chiefari, J. Controlled Radical Polymerization; American Chemical Society: Washington, DC, USA, 2016. [Google Scholar]
- Ballard, N.; Mecerreyes, D.; Asua, J.M. Redox active compounds in controlled radical polymerization and dye-sensitized solar cells: Mutual solutions to disparate problems. Chem. A Eur. J. 2015, 21, 18516–18527. [Google Scholar] [CrossRef] [PubMed]
- Mastan, E.; Li, X.; Zhu, S. Modeling and theoretical development in controlled radical polymerization. Prog. Polym. Sci. 2015, 45, 71–101. [Google Scholar] [CrossRef]
- Chan, N.; Cunningham, M.F.; Hutchinson, R.A. Copper-mediated controlled radical polymerization in continuous flow processes: Synergy between polymer reaction engineering and innovative chemistry. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3081–3096. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Delaittre, G.; Gruendling, T.; Paulöhrl, T. Elucidation of reaction mechanisms and polymer structure: Living/controlled radical polymerization. In Mass Spectrometry in Polymer Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 373–403. [Google Scholar]
- Flores, J.D.; Abel, B.A.; Smith, D.; McCormick, C.L. Stimuli-responsive polymers via controlled radical polymerization. In Monitoring Polymerization Reactions; John Wiley & Sons: New York, NY, USA, 2013; pp. 45–58. [Google Scholar]
- Hemp, S.T.; Long, T.E. DNA-inspired hierarchical polymer design: Electrostatics and hydrogen bonding in concert. Macromol. Biosci. 2012, 12, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zhang, M.; Dixit, N.; Moore, R.B.; Long, T.E. Nucleobase self-assembly in supramolecular adhesives. Macromolecules 2012, 45, 805–812. [Google Scholar] [CrossRef]
- Roy, R.K.; Meszynska, A.; Laure, C.; Charles, L.; Verchin, C.; Lutz, J.-F. Design and synthesis of digitally encoded polymers that can be decoded and erased. Nat. Commun. 2015, 6, 7237. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F.; Ouchi, M.; Liu, D.R.; Sawamoto, M. Sequence-controlled polymers. Science 2013, 341, 1238149. [Google Scholar] [CrossRef] [PubMed]
- Landau, L.D.; Lifshitz, E.M. Statistical Physics, 3rd ed.; Pergammon Press: Oxford, UK; New York, NY, USA, 1980. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: New York, NY, USA, 1953. [Google Scholar]
- Yamakawa, H. Modern Theory of Polymer Solutions; Harper & Row: New York, NY, USA, 1971. [Google Scholar]
- McMillan, W.G., Jr.; Mayer, J.E. The statistical thermodynamics of multicomponent systems. J. Chem. Phys. 1945, 13, 276–305. [Google Scholar] [CrossRef]
- Hansen, J.-P.; McDonald, I.R. Theory of Simple Liquids; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- Odijk, T. Polyelectrolytes near the rod limit. J. Polymer Sci. Polym. Phys. Ed. 1977, 15, 477–483. [Google Scholar] [CrossRef]
- Skolnick, J.; Fixman, M. Electrostatic persistence length of a wormlike polyelectrolyte. Macromolecules 1977, 10, 944–948. [Google Scholar] [CrossRef]
- Sorci, G.A.; Reed, W.F. Electrostatically enhanced second and third virial coefficients, viscosity, and interparticle correlations for linear polyelectrolytes. Macromolecules 2002, 35, 5218–5227. [Google Scholar] [CrossRef]
- Reed, W.; Ghosh, S.; Medjahdi, G.; Francois, J. Dependence of polyelectrolyte apparent persistence lengths, viscosity, and diffusion on ionic strength and linear charge density. Macromolecules 1991, 24, 6189–6198. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte solutions II. Self-diffusion of the small ions. J. Chem. Phys. 1969, 51, 934–938. [Google Scholar] [CrossRef]
- Oosawa, F. Polyelectrolytes; Marcel Dekker Inc.: New York, NY, USA, 1971. [Google Scholar]
- Wilson, R.W.; Bloomfield, V.A. Counterion-induced condesation of deoxyribonucleic acid. A light-scattering study. Biochemistry 1979, 18, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Hinderberger, D.; Spiess, H.W.; Jeschke, G. Dynamics, site binding, and distribution of counterions in polyelectrolyte solutions studied by electron paramagnetic resonance spectroscopy. J. Phys. Chem. B 2004, 108, 3698–3704. [Google Scholar] [CrossRef]
- Kreft, T.; Reed, W.F. Experimental observation of crossover from noncondensed to counterion condensed regimes during free radical polyelectrolyte copolymerization under high-composition drift conditions. J. Phys. Chem. B 2009, 113, 8303–8309. [Google Scholar] [CrossRef] [PubMed]
- Mayo, F.R.; Lewis, F.M. Copolymerization. I. A basis for comparing the behavior of monomers in copolymerization; the copolymerization of styrene and methyl methacrylate. J. Am. Chem. Soc. 1944, 66, 1594–1601. [Google Scholar] [CrossRef]
- Kreft, T.; Reed, W.F. Predictive control of average composition and molecular weight distributions in semibatch free radical copolymerization reactions. Macromolecules 2009, 42, 5558–5565. [Google Scholar] [CrossRef]
- Florenzano, F.H.; Strelitzki, R.; Reed, W.F. Absolute, on-line monitoring of molar mass during polymerization reactions. Macromolecules 1998, 31, 7226–7238. [Google Scholar] [CrossRef]
- Reed, W.F. Automated continuous online monitoring of polymerization reactions (acomp) and related techniques. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: New York, NY, USA, 2006. [Google Scholar]
- Çatalgil-Giz, H.; Giz, A.; Alb, A.M.; Öncül Koç, A.; Reed, W.F. Online monitoring of composition, sequence length, and molecular weight distributions during free radical copolymerization, and subsequent determination of reactivity ratios. Macromolecules 2002, 35, 6557–6571. [Google Scholar] [CrossRef]
- Alb, A.M.; Enohnyaket, P.; Drenski, M.F.; Head, A.; Reed, A.W.; Reed, W.F. Online monitoring of copolymerization involving comonomers of similar spectral characteristics. Macromolecules 2006, 39, 5705–5713. [Google Scholar] [CrossRef]
- Alb, A.M.; Paril, A.; Catalfil-Giz, H.; Giz, A.; Reed, W.F. Evolution of composition, molar mass, and conductivity during the free radical copolymerization or polyelectrolytes. J. Phys. Chem. B 2007, 111, 8560–8566. [Google Scholar] [CrossRef] [PubMed]
- Farinato, R.S.; Calbick, J.; Sorci, G.A.; Florenzano, F.H.; Reed, W.F. Online monitoring of the final, divergent growth phase in the step-growth polymerization of polyamines. Macromolecules 2005, 38, 1148–1158. [Google Scholar] [CrossRef]
- Alb, A.M.; Reed, W.F. Simultaneous monitoring of polymer and particle characteristics during emulsion polymerization. Macromolecules 2008, 41, 2406–2414. [Google Scholar] [CrossRef]
- Alb, A.M.; Farinato, R.; Calbick, J.; Reed, W.F. Online monitoring of polymerization reactions in inverse emulsions. Langmuir 2006, 22, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Kreft, T.; Reed, W.F. Predictive control and verification of conversion kinetics and polymer molecular weight in semi-batch free radical homopolymer reactions. Eur. Polym. J. 2009, 45, 2288–2303. [Google Scholar] [CrossRef]
- Grassl, B.; Reed, W.F. Online polymerization monitoring in a continuous reactor. Macromol. Chem. Phys. 2002, 203, 586–597. [Google Scholar] [CrossRef]
- Paril, A.; Alb, A.M.; Reed, W.F. Online monitoring of the evolution of polyelectrolyte characteristics during postpolymerization modification processes. Macromolecules 2007, 40, 4409–4413. [Google Scholar] [CrossRef]
- Mignard, E.; Leblanc, T.; Bertin, D.; Guerret, O.; Reed, W.F. Online monitoring of controlled radical polymerization: Nitroxide-mediated gradient copolymerization. Macromolecules 2004, 37, 966–975. [Google Scholar] [CrossRef]
- Mignard, E.; Lutz, J.-F.; Leblanc, T.; Matyjaszewski, K.; Guerret, O.; Reed, W.F. Kinetics and molar mass evolution during atom transfer radical polymerization of n-butyl acrylate using automatic continuous online monitoring. Macromolecules 2005, 38, 9556–9563. [Google Scholar] [CrossRef]
- Alb, A.M.; Serelis, A.K.; Reed, W.F. Kinetic trends in raft homopolymerization from online monitoring. Macromolecules 2008, 41, 332–338. [Google Scholar] [CrossRef]
- Alb, A.M.; Enohnyaket, P.; Craymer, J.F.; Eren, T.; Coughlin, E.B.; Reed, W.F. Online monitoring of ring-opening metathesis polymerization of cyclooctadiene and a functionalized norbornene. Macromolecules 2007, 40, 444–451. [Google Scholar] [CrossRef]
- McAfee, T.; Leonardi, N.; Montgomery, R.; Siqueira, J.; Zekoski, T.; Drenski, M.F.; Reed, W.F. Automatic control of polymer molecular weight during synthesis. Macromolecules 2016, 49, 7170–7183. [Google Scholar] [CrossRef]
- Alb, A.M.; Drenski, M.F.; Reed, W.F. Simultaneous continuous, nonchromatographic monitoring and discrete chromatographic monitoring of polymerization reactions. J. Appl. Polym. Sci. 2009, 113, 190–198. [Google Scholar] [CrossRef]
- McFaul, C.A.; Alb, A.M.; Drenski, M.F.; Reed, W.F. Simultaneous multiple sample light scattering detection of LCST during copolymer synthesis. Polymer 2011, 52, 4825–4833. [Google Scholar] [CrossRef]
- McFaul, C.A.; Drenski, M.F.; Reed, W.F. Online, continuous monitoring of the sensitivity of the LCST of nipam-Am copolymers to discrete and broad composition distributions. Polymer 2014, 55, 4899–4907. [Google Scholar] [CrossRef]
- Huggins, M.L. The viscosity of dilute solutions of long-chain molecules. J. Am. Chem. Soc. 1942, 64, 2716–2718. [Google Scholar] [CrossRef]
- Zimm, B.; Stein, R.; Doty, P. Polymer bull. 1 (1945) 90; bh zimm. J. Chem. Phys. 1948, 16, 1093. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids; Acdamic Press: New York, NY, USA, 1986. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Type | Am0 (g/cm3) | SSo (g/cm3) | Initiator (g/cm3) | Second Dilution | First Dilution | T (°C) | Figures |
|---|---|---|---|---|---|---|---|---|
| A | 50/50 batch | 12.9 × 10−3 | 37.5 × 10−3 | 2.73 × 10−3 | 50× | 10× | 65 | Figure 1a,b, Figure 2, Figure 3, Figure 7a |
| B | 50/50 batch | 10.3 × 10−3 | 32.5 × 10−3 | 5.00 × 10−4 | 105× | 15× | 60 | Figure 4a,b, Figure 5, Figure 6 |
| C | Semi-batch | 18.1 × 10−3 | 181 × 10−3 stock fed into reactor at 0.1 mL/min | 2.73 × 10−3 | 50× | 10× | 65 | Figure 7b |
| Reaction A | Reaction B | Reaction C (Semi-Batch) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Stage | [NaCl] * (mM) | IS (mM) | Dilution factor | [NaCl] * (mM) | IS (mM) | Dilution factor | [NaCl] * (mM) | IS (mM) | Dilution factor |
| 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 |
| 2 | 2 | 0.095 | 1.05 | 4 | 0.12 | 1.03125 | 2 | 0.095 | 1.05 |
| 3 | 20 | 1 | 1.10 | 40 | 1.3 | 1.0625 | 20 | 1 | 1.10 |
| 4 | 200 | 9.7 | 1.15 | 400 | 13 | 1.09375 | 200 | 9.7 | 1.15 |
| 5 | 1000 | 51 | 1.20 | 5000 | 151 | 1.125 | 1000 | 51 | 1.20 |
| 6 | 2000 | 129 | 1.25 | - | - | - | 2000 | 129 | 1.25 |
| 7 | 5000 | 316 | 1.30 | - | - | - | 5000 | 316 | 1.30 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, A.; Zhu, Z.; Drenski, M.F.; Reed, W.F. Simultaneous Monitoring of the Effects of Multiple Ionic Strengths on Properties of Copolymeric Polyelectrolytes during Their Synthesis. Processes 2017, 5, 17. https://doi.org/10.3390/pr5020017
Wu A, Zhu Z, Drenski MF, Reed WF. Simultaneous Monitoring of the Effects of Multiple Ionic Strengths on Properties of Copolymeric Polyelectrolytes during Their Synthesis. Processes. 2017; 5(2):17. https://doi.org/10.3390/pr5020017
Chicago/Turabian StyleWu, Aide, Zifu Zhu, Michael F. Drenski, and Wayne F. Reed. 2017. "Simultaneous Monitoring of the Effects of Multiple Ionic Strengths on Properties of Copolymeric Polyelectrolytes during Their Synthesis" Processes 5, no. 2: 17. https://doi.org/10.3390/pr5020017
APA StyleWu, A., Zhu, Z., Drenski, M. F., & Reed, W. F. (2017). Simultaneous Monitoring of the Effects of Multiple Ionic Strengths on Properties of Copolymeric Polyelectrolytes during Their Synthesis. Processes, 5(2), 17. https://doi.org/10.3390/pr5020017