Characterizing Gene and Protein Crosstalks in Subjects at Risk of Developing Alzheimer’s Disease: A New Computational Approach


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
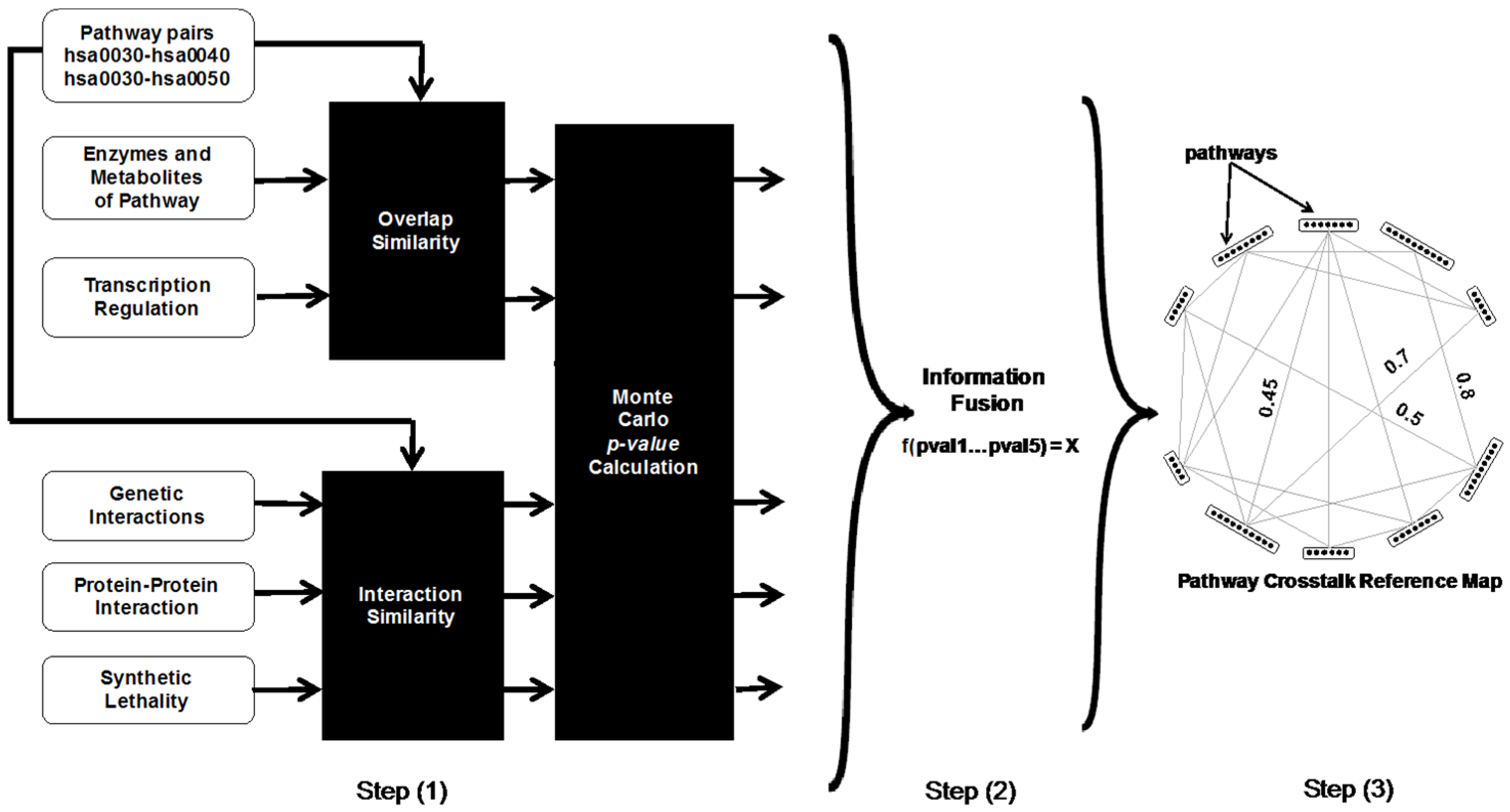
2.1. Identification of Potential Pathway Crosstalks
2.1.1. Scoring Pathway Crosstalks Based on Common Elements
- Shared enzymes and metabolites: The number of enzymes and metabolites shared by a pair of pathways is utilized as one of the evidence to identify potential pathway crosstalks. This is reasonable because a variation in the concentration of common enzymes or metabolites will affect both pathways.
- Phosphorylation: Phosphorylation, performed by protein kinases, is the addition of a phosphate group to a protein, which results in a change of the protein’s function. Co-phosphorylated proteins in different pathways suggest potential pathway crosstalks.
- Transcriptional regulation: Genes with common transcription factors are likely coexpressed. Coexpressed genes in different pathways provide an avenue for the pathways to crosstalk. For each pathway pair, we find the group of transcription factors that have coexpressed genes in both pathways.
2.1.2. Scoring Pathway Crosstalks Based on Interacting Elements
- Genetic interactions: The use of genetic interactions for identifying pathway crosstalks stems from the concept of “between-pathway” interactions. This essentially states that if there is a genetic interaction between pathways, one pathway covers for the defects in the other pathway.
- Protein domain: Protein function is closely related to fundamental units of protein structure called “domains”. In the domain interaction network, a pair of proteins has an edge if they are associated with the same set of protein domains. These edges are taken into consideration to assess for potential pathway crosstalks because of the common domains.
2.1.3. Significance Estimation of Pathway Crosstalk Scores
- For each pair of pathways, a score for how likely they are to crosstalk is calculated based on each evidence.
- Each pathway is randomized by replacing all proteins in that pathway with randomly selected proteins from the set of all proteins in the organism. This pathway randomization step is repeated W = 1000 times, i.e., we obtain W sets of pathways with randomized proteins.
- The evidence-specific scores for each pathway pair are recalculated W times using each set of pathways with randomized proteins.
- An evidence-specific p-value is estimated for each pathway pair as R/W, where R is the number of randomized versions of that pathway pair that produce an evidence-specific score greater than or equal to the score obtained for the original pathway pair.
2.1.4. Combining the Scores for Each Pathway Crosstalk
2.2. Identification of Patient-Specific Pathway Crosstalks
- Obtain a mapping of SNPs to pathways using genetic information.
- Identify the list of SNPs that are present in a patient.
- Use the mapping obtained in Step 1 and the patient-specific SNP list in Step 2 to obtain the pathways that are “SNP-enriched” in the patient.
- Use the “SNP-enriched” pathways from Step 3 to obtain patient-specific pathway crosstalks.
2.2.1. Obtain a Mapping of SNPs to Pathways
2.2.2. Identify Patient-Specific SNPs That Are Present
2.2.3. Identify Patient-Specific SNP-Enriched Pathways
2.2.4. Identify Patient-Specific Pathway Crosstalks
2.3. Identification of Biased Pathway Crosstalk
- Population: n is the total number of patients.
- Success in population: x is the total number of MCI progressive patients and is the number of MCI non-progressive patients.
- Sample: is the total number of patients (both MCI progressive and MCI non-progressive patients) a pathway crosstalk is enriched in.
- Success in sample: is the number of MCI progressive patients and is the number of MCI non-progressive patients the pathway crosstalk is enriched in.
2.4. Datasets
3. Results/Discussion
3.1. Sample Characteristics
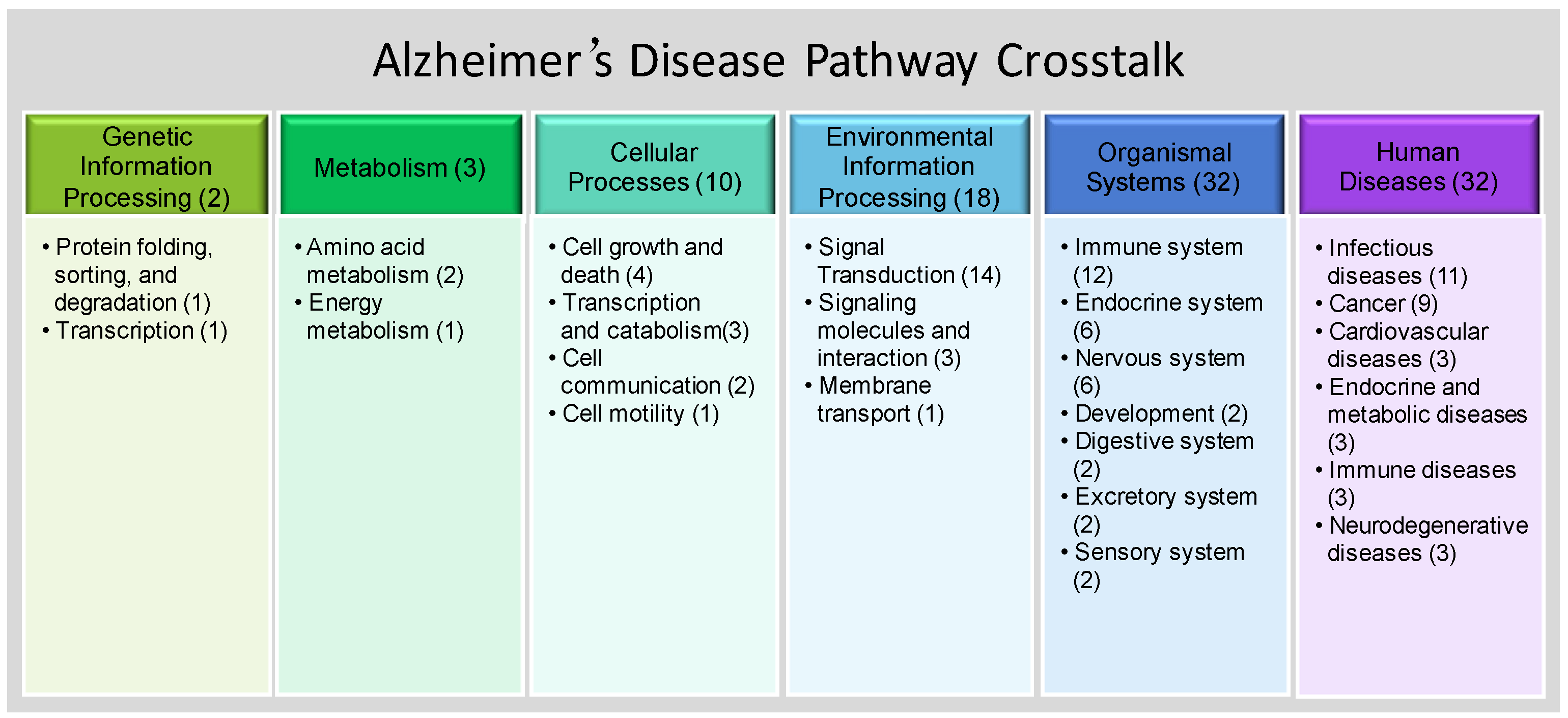
3.2. SNP-Enriched Pathways and Associated Crosstalks
3.3. SNP-Enriched Features with Baseline Clinical Parameters
3.4. Comparison of Model Performances from Shaffer et al. (2013) with Our Model Performance including SNP-Enriched Features
3.5. Randomized SNP-Enriched Features
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brookmeyer, R.; Evans, D.A.; Hebert, L.; Langa, K.M.; Heeringa, S.G.; Plassman, B.L.; Kukull, W.A. National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement. 2011, 7, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2017 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2017, 13, 325–373. [Google Scholar]
- Heron, M. Deaths: Leading causes for 2010. Natl. Vital Stat. Rep. 2013, 62, 1–96. [Google Scholar] [PubMed]
- Saykin, A.J.; Shen, L.; Yao, X.; Kim, S.; Nho, K.; Risacher, S.L.; Ramanan, V.K.; Foroud, T.M.; Faber, K.M.; Sarwar, N.; et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: Progress, opportunities, and plans. Alzheimers Dement. 2015, 11, 792–814. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Tsivinsky, V.G.; Abdullah, L.; Crawford, F.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment: Biomarker validation study. Aging Albany N.Y. 2013, 5, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. Alzheimers Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef]
- Femminella, G.D.; Ferrara, N.; Rengo, G. The emerging role of microRNAs in Alzheimer’s disease. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Padmanabhan, K.; Rocha, A.M.; Shpanskaya, Y.; Mihelcic, J.R.; Scott, K.; Samatova, N.F. SPICE: Discovery of phenotype-determining component interplays. BMC Syst. Biol. 2012, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.S.; Gianoulis, T.A.; Liu, Y.; Li, J.; Paccanaro, A.; Lussier, Y.A.; Gerstein, M. Integration of curated databases to identify genotype-phenotype associations. BMC Genom. 2006, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.; Zimmer, R. Assigning functional linkages to proteins using phylogenetic profiles and continuous phenotypes. Bioinformatics 2008, 24, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, W.; Rocha, A.M.; Elmore, M.T.; Trien, J.; Samatova, N.F. Discovery of Enriched Biological Motifs Using Knowledge Priors with Application to Biohydrogen Production. In Proceedings of the BIOCOMP, Las Vegas, NV, USA, 12–15 July 2010; pp. 17–23. [Google Scholar]
- Hendrix, W.; Rocha, A.M.; Padmanabhan, K.; Choudhary, A.; Scott, K.; Mihelcic, J.R.; Samatova, N.F. DENSE: Efficient and prior knowledge-driven discovery of phenotype associated protein functional modules. BMC Syst. Biol. 2011, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Jim, K.; Parmar, K.; Singh, M.; Tavazoie, S. A cross-genomic approach for systematic mapping of phenotypic traits to genes. Genom. Res. 2004, 14, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.C.; Rocha, A.M.; Padmanabhan, K.; Chen, Z.; Scott, K.; Mihelcic, J.R.; Samatova, N.F. Efficient α,β-motif finder for identification of phenotype-related functional modules. BMC Bioinform. 2011, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.C.; Rocha, A.M.; Padmanabhan, K.; Shpanskaya, Y.; Banfield, J.; Scott, K.; Mihelcic, J.R.; Samatova, N.F. NIBBS-Search for fast and accurate prediction of phenotype-biased metabolic systems. PLoS Comput. Biol. 2012, 8, e1002490. [Google Scholar] [CrossRef] [PubMed]
- Slonim, N.; Elemento, O.; Tavazoie, S. Ab initio genotype-phenotype association reveals intrinsic modularity in genetic networks. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Doerks, T.; Jensen, L.J.; Perez-Iratxeta, C.; Kaczanowski, S.; Hooper, S.D.; Andrade, M.A.; Bork, P. Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol. 2005, 3, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, M.; Shasha, D.; Kim, W.; Surette, M.G.; Benfey, P.N. Trait-to-Gene: A computational method for predicting the function of uncharacterized genes. Curr. Biol. 2003, 13, 129–133. [Google Scholar] [CrossRef]
- Tamura, M.; D’haeseleer, P. Microbial genotype-phenotype mapping by class association rule mining. Bioinformatics 2008, 24, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Zalesky, A.; Fornito, A.; Bullmore, E.T. Network-based statistic: Identifying differences in brain networks. Neuroimage 2010, 53, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Uberti, D.; Racchi, M.; Govoni, S.; Memo, M. Unfolded p53: A potential biomarker for Alzheimer’s disease. J. Alzheimers Dis. 2007, 12, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Agarwal, P.; Rajagopalan, D. A global pathway crosstalk network. Bioinformatics 2008, 24, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Godoy Zeballos, J.A. Signaling pathway cross talk in Alzheimer’s disease. Cell Commun. Signal. 2014, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 2, 145–175. [Google Scholar] [PubMed]
- Myers, C.L.; Robson, D.; Wible, A.; Hibbs, M.A.; Chiriac, C.; Theesfeld, C.L.; Dolinski, K.; Troyanskaya, O.G. Discovery of biological networks from diverse functional genomic data. Genome Biol. 2005, 6, R114. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, W.; Chang, Z.; Duanmu, H.; Zhang, S.; Li, Z.; Yu, L.; Li, X. Prediction of human protein-protein interaction by a mixed Bayesian model and its application to exploring underlying cancer-related pathway crosstalk. J. R. Soc. Interface 2011, 8, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wang, Y.; Zhang, X.S.; Chen, L. Identifying dysfunctional crosstalk of pathways in various regions of Alzheimer’s disease brains. BMC Syst. Biol. 2010, 4, S11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Pathway crosstalk network. In Systems Biology for Signaling Networks; Choi, S., Ed.; Springer: New York, NY, USA, 2010; pp. 491–504. [Google Scholar]
- Hartman, J.L.; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Suthers, P.F.; Zomorrodi, A.; Maranas, C.D. Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 2009, 5, 301. [Google Scholar] [CrossRef] [PubMed]
- North, B.V.; Curtis, D.; Sham, P.C. A note on the calculation of empirical p-values from Monte Carlo procedures. Am. J. Hum. Genet. 2002, 71, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Feller, W. An Introduction to Probability Theory and Its Applications; Wiley: New York, NY, USA, 1957. [Google Scholar]
- Silver, M.; Janousova, E.; Hua, X.; Thompson, P.M.; Montana, G. Identification of gene pathways implicated in Alzheimer’s disease using longitudinal imaging phenotypes with sparse regression. Neuroimage 2012, 63, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.; Montana, G. Fast identification of biological pathways associated with a quantitative trait using group lasso with overlaps. Stat. Appl. Genet. Mol. Biol. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2005, 34, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, H.; Severin, J.; Lizio, M.; Forrest, A.R.; van Nimwegen, E.; Rehli, M.; Schroder, K.; Irvine, K.; Suzuki, H.; Carninci, P.; et al. Update of the FANTOM web resource: From mammalian transcriptional landscape to its dynamic regulation. Nucleic Acids Res. 2010, 39, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, H.; Severin, J.; Lizio, M.; Waterhouse, A.; Katayama, S.; Irvine, K.M.; Hume, D.A.; Forrest, A.R.R.; Suzuki, H.; Carninci, P.; Hayashizaki, Y.; Daub, C.O. The FANTOM web resource: From mammalian transcriptional landscape to its dynamic regulation. Genome Biol. 2009, 10, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Du, P.; Xu, N. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PLoS ONE 2010, 5, e15411. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Murphy, C.G.; Johnson, R.; Lay, J.M.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Rosenstein, M.C.; Wiegers, T.C.; et al. The comparative toxicogenomics database: Update 2013. Nucleic Acids Res. 2013, 41, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Cariaso, M.; Lennon, G. SNPedia: A wiki supporting personal genome annotation, interpretation and analysis. Nucleic Acids Res. 2012, 40, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, S. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Wiener, H.W.; Perry, R.T.; Chen, Z.; Harrell, L.E.; Go, R.C. A polymorphism in SOD2 is associated with development of Alzheimer’s disease. Genes Brain Behav. 2007, 6, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Dahiyat, M.; Cumming, A.; Harrington, C.; Wischik, C.; Xuereb, J.; Corrigan, F.; Breen, G.; Shaw, D.; St Clair, D. Association between Alzheimer’s disease and the NOS3 gene. Ann. Neurol. 1999, 46, 664–667. [Google Scholar] [CrossRef]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; Liu, E.; et al. The Alzheimer’s disease neuroimaging initiative: A review of papers published since its inception. Alzheimers Dement. 2012, 8, 1–68. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.L.; Petrella, J.R.; Sheldon, F.C.; Choudhury, K.R.; Calhoun, V.D.; Coleman, R.E.; Doraiswamy, P.M. Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology 2013, 266, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Vignini, A.; Giulietti, A.; Nanetti, L.; Raffaelli, F.; Giusti, L.; Mazzanti, L.; Provinciali, L. Alzheimer’s disease and diabetes: New insights and unifying therapies. Curr. Diabetes Rev. 2013, 9, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Kim, S.; Holohan, K.; Shen, L.; Nho, K.; Risacher, S.L.; Foroud, T.M.; Mukherjee, S.; Crane, P.K.; Aisen, P.S.; et al. Genome-wide pathway analysis of memory impairment in the Alzheimer’s disease neuroimaging initiative (ADNI) cohort implicates gene candidates, canonical pathways, and networks. Brain Imaging Behav. 2012, 6, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.M.; Behrens, M.I.; Xiong, C.; Miller, J.P.; Morris, J.C. Alzheimer disease and cancer. Neurology 2005, 64, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Honjo, K.; van Reekum, R.; Verhoeff, N.P. Alzheimer’s disease and infection: Do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 2009, 5, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.K.; Pyo, K.H.; Shin, K.Y.; Hwang, Y.S.; Lim, H.; Lee, S.J.; Moon, J.H.; Lee, S.H.; Suh, Y.H.; Chai, J.Y.; et al. Toxoplasma gondii infection in the brain inhibits neuronal degeneration and learning and memory impairments in a murine model of Alzheimer’s disease. PLoS ONE 2012, 7, e33312. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansoori, K.M.; Hasan, M.Y.; Al-Hayani, A.; El-Agnaf, O.M. The role of α-synuclein in neurodegenerative diseases: From molecular pathways in disease to therapeutic approaches. Curr. Alzheimer Res. 2013, 10, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef] [PubMed]
- Berntorp, K.; Frid, A.; Alm, R.; Fredrikson, G.N.; Sjöberg, K.; Ohlsson, B. Antibodies against gonadotropin-releasing hormone (GnRH) in patients with diabetes mellitus is associated with lower body weight and autonomic neuropathy. BMC Res. Notes 2013, 6, 329. [Google Scholar] [CrossRef] [PubMed]
- Giunta, B.; Fernandez, F.; Nikolic, W.V.; Obregon, D.; Rrapo, E.; Town, T.; Tan, J. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Zeng, F.; Wang, Y.R.; Zhou, H.D.; Giunta, B.; Tan, J.; Wang, Y.J. Immunity and Alzheimer’s disease: Immunological perspectives on the development of novel therapies. Drug Discov. Today 2013, 18, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Freo, U.; Pizzolato, G.; Dam, M.; Ori, C.; Battistin, L. A short review of cognitive and functional neuroimaging studies of cholinergic drugs: Implications for therapeutic potentials. J. Neural Transm. 2002, 109, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, A.; Dudilot, A.; Licea, S.; Bourgeois, C.; Boehm, J. NMDA-Receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS ONE 2013, 8, e65350. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.S.; Wei, W.Z.; Shimahara, T.; Xie, C.W. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol. Learn. Mem. 2002, 77, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Ghebranious, N.; Mukesh, B.; Giampietro, P.F.; Glurich, I.; Mickel, S.F.; Waring, S.C.; Mc-Carty, C.A. A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin. Med. Res. 2011, 9, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. The DNA replication stress hypothesis of Alzheimer’s disease. Sci. World J. 2011, 11, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [PubMed]
- Bradley, W.G.; Polinsky, R.J.; Pendlebury, W.W.; Jones, S.K.; Nee, L.E.; Bartlett, J.D.; Hartshorn, J.N.; Tandan, R.; Sweet, L.; Magin, G.K. DNA repair deficiency for alkylation damage in cells from Alzheimer’s disease patients. Prog. Clin. Biol. Res. 1989, 317, 715–732. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Evidence |
|---|---|
| APP amyloid beta (A4) precursor protein | Mutations in this gene have been implicated in autosomal dominant AD and cerebroarterial amyloidosis (NCBI Entrez Gene) |
| IL-1β | Four new genetic studies underscore the relevance of IL-1 to Alzheimer’s pathogenesis, showing that homozygosity of a specific polymorphism in the IL-1α gene at least triples Alzheimer’s risk, especially for an earlier age of onset and in combination with homozygosity for another polymorphism in the IL-1β gene [48] |
| SOD2 | A polymorphism in SOD2 is associated with development of AD [49] |
| NOS3 | NOS3 may be a new genetic risk factor of late onset AD [50] |
| Subjects with MCI (n = 91) | MCI Progressive Patients (n = 41) | MCI Non-Progressive Patients (n = 50) | p-Value 1 |
|---|---|---|---|
| Age (years) | 75.17 ± 7.30 | 74.78 ± 7.44 | 0.8011 |
| Male-to-female-ratio 2,3 | 2.42 (29/12) | 2.33 (35/15) | 0.9394 |
| Family history of AD 2,3 | 36.39 (15/41) | 36.00 (18/50) | 0.9539 |
| APOE ε4 carriers, % 2,3 | 60.98 (25/41) | 50.00 (25/50) | 0.4036 |
| Average follow-up time (months) | 34.10 ± 9.70 | 29.64 ± 10.94 | 0.0426 |
| Metrics | Baseline Clinical: Age, Education, ADAS-Cog | Significant Pathways (Only) | Significant Pathway Crosstalks (Only) | Baseline Clinical + Significant Pathways | Baseline Clinical + Significant Pathway Crosstalks |
|---|---|---|---|---|---|
| Accuracy in % | 59.19 ± 2.46 | 56.78 ± 3.5 | 60.97 ± 3.24 | 64.57 ± 3.56 | 70.9 ± 3.3 |
| Support Vectors in % | 83.64 ± 0.29 | 68.36 ± 2.1 | 50.83 ± 4.77 | 63.3 ± 1.15 | 54.29 ± 0.56 |
| True Positives | 30.78 ± 1.7 | 31.21 ± 4.7 | 40.9 ± 3.1 | 33.64 ± 2.4 | 37.93 ± 2.16 |
| False Negatives | 19.22 ± 1.7 | 18.79 ± 4.7 | 9.06 ± 3.13 | 16.36 ± 2.4 | 12.07 ± 2.16 |
| False Positives | 17.9 ± 1.6 | 20.51 ± 3.09 | 26.46 ± 4.17 | 15.9 ± 2.03 | 14.41 ± 1.6 |
| True Negatives | 23.08 ± 1.55 | 20.49 ± 3.09 | 14.54 ± 4.17 | 25.11 ± 2.03 | 26.59 ± 1.6 |
| Sensitivity | 0.62 ± 0.03 | 0.62 ± 0.09 | 0.82 ± 0.06 | 0.67 ± 0.05 | 0.76 ± 0.04 |
| Specificity | 0.56 ± 0.04 | 0.51 ± 0.07 | 0.35 ± 0.11 | 0.61 ± 0.05 | 0.65 ± 0.04 |
| Precision | 0.62 ± 0.03 | 0.60 ± 0.09 | 0.61 ± 0.03 | 0.68 ± 0.03 | 0.74 ± 0.03 |
| Model | Logistic Regression | SVM with Linear Kernel | ||||
|---|---|---|---|---|---|---|
| No. Patient | 97 Patients | 91 Patients | 91 Patients | |||
| Metrics | original 10-fold cross-validation: Baseline Clinical + APOE ε4 Shaffer et al. paper [52] | 100 Iterations of Random 10-fold cross-validation: Baseline Clinical + APOE ε4 | 100 Iterations of Random 10-fold cross-validation: Baseline Clinical + APOE ε4 | 100 Iterations of Random 10-fold cross-validation: Baseline Clinical + APOE ε4 + significant pathway crosstalks | 100 Iterations of Random 10-fold cross-validation: Baseline Clinical + APOE ε4 + significant pathway | 100 Iterations of Random 10-fold cross-validation: Baseline Clinical + APOE ε4 + significant pathway crosstalks |
| Accuracy in % | 58.7 | 59.10 ± 1.71 | 57.04 ± 2 | 72.1 ± 2.66 | 63.56 ± 3.4 | 69.53 ± 2.9 |
| Support Vectors in % | N/A | N/A | N/A | N/A | 63.29 ± 1.16 | 53.65 ± 0.6 |
| True Positives | 17 | 39.98 ± 1.21 | 16.48 ± 1.16 | 39.22 ± 2.08 | 33.74 ± 2.4 | 37.6 ± 1.9 |
| False Negatives | 26 | 14.02 ± 1.21 | 24.43 ± 1.3 | 10.78 ± 2.08 | 16.3 ± 2.35 | 12.44 ± 1.9 |
| False Positives | 14 | 25.65 ± 1.23 | 14.67 ± 1.58 | 14.61 ± 1.3 | 16.91 ± 2 | 15.29 ± 1.5 |
| True Negatives | 40 | 17.35 ± 1.23 | 35.52 ± 1.53 | 26.39 ± 1.3 | 24.09 ± 2 | 25.71 ± 1.5 |
| Sensitivity | 0.40 | 0.74 ± 0.02 | 0.41 ± 0.05 | 0.78 ± 0.04 | 0.68 ± 0.05 | 0.75 ± 0.04 |
| Specificity | 0.74 | 0.40 ± 0.03 | 0.72 ± 0.03 | 0.64 ± 0.03 | 0.59 ± 0.05 | 0.63 ± 0.04 |
| Precision | 0.46 | 0.61 ± 0.02 | 0.54 ± 0.06 | 0.75 ± 0.03 | 0.68 ± 0.04 | 0.73 ± 0.03 |
| Metrics | Baseline Clinical + Randomized Significant Pathway Crosstalks | Baseline Clinical + Significant Pathway Crosstalks |
|---|---|---|
| Accuracy in % | 59.27 ± 3.66 | 70.9 ± 3.3 |
| Support Vectors in % | 83.47 ± 1.84 | 54.29 ± 0.56 |
| True Positives | 30.86 ± 1.98 | 37.93 ± 2.16 |
| False Negatives | 19.14 ± 1.97 | 12.07 ± 2.16 |
| False Positives | 17.95 ± 1.59 | 14.41 ± 1.6 |
| True Negatives | 23.05 ± 1.56 | 26.59 ± 1.6 |
| Sensitivity | 0.62 ± 0.97 | 0.76 ± 0.04 |
| Specificity | 0.56 ± 1.45 | 0.65 ± 0.04 |
| Precision | 0.63 ± 0.02 | 0.74 ± 0.03 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padmanabhan, K.; Nudelman, K.; Harenberg, S.; Bello, G.; Sohn, D.; Shpanskaya, K.; Tiwari Dikshit, P.; Yerramsetty, P.S.; Tanzi, R.E.; Saykin, A.J.; et al. Characterizing Gene and Protein Crosstalks in Subjects at Risk of Developing Alzheimer’s Disease: A New Computational Approach. Processes 2017, 5, 47. https://doi.org/10.3390/pr5030047
Padmanabhan K, Nudelman K, Harenberg S, Bello G, Sohn D, Shpanskaya K, Tiwari Dikshit P, Yerramsetty PS, Tanzi RE, Saykin AJ, et al. Characterizing Gene and Protein Crosstalks in Subjects at Risk of Developing Alzheimer’s Disease: A New Computational Approach. Processes. 2017; 5(3):47. https://doi.org/10.3390/pr5030047
Chicago/Turabian StylePadmanabhan, Kanchana, Kelly Nudelman, Steve Harenberg, Gonzalo Bello, Dongwha Sohn, Katie Shpanskaya, Priyanka Tiwari Dikshit, Pallavi S. Yerramsetty, Rudolph E. Tanzi, Andrew J. Saykin, and et al. 2017. "Characterizing Gene and Protein Crosstalks in Subjects at Risk of Developing Alzheimer’s Disease: A New Computational Approach" Processes 5, no. 3: 47. https://doi.org/10.3390/pr5030047
APA StylePadmanabhan, K., Nudelman, K., Harenberg, S., Bello, G., Sohn, D., Shpanskaya, K., Tiwari Dikshit, P., Yerramsetty, P. S., Tanzi, R. E., Saykin, A. J., Petrella, J. R., Doraiswamy, P. M., Samatova, N. F., & Alzheimer’s Disease Neuroimaging Initiative. (2017). Characterizing Gene and Protein Crosstalks in Subjects at Risk of Developing Alzheimer’s Disease: A New Computational Approach. Processes, 5(3), 47. https://doi.org/10.3390/pr5030047