Direct Solid Oxide Electrolysis of Carbon Dioxide: Analysis of Performance and Processes

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
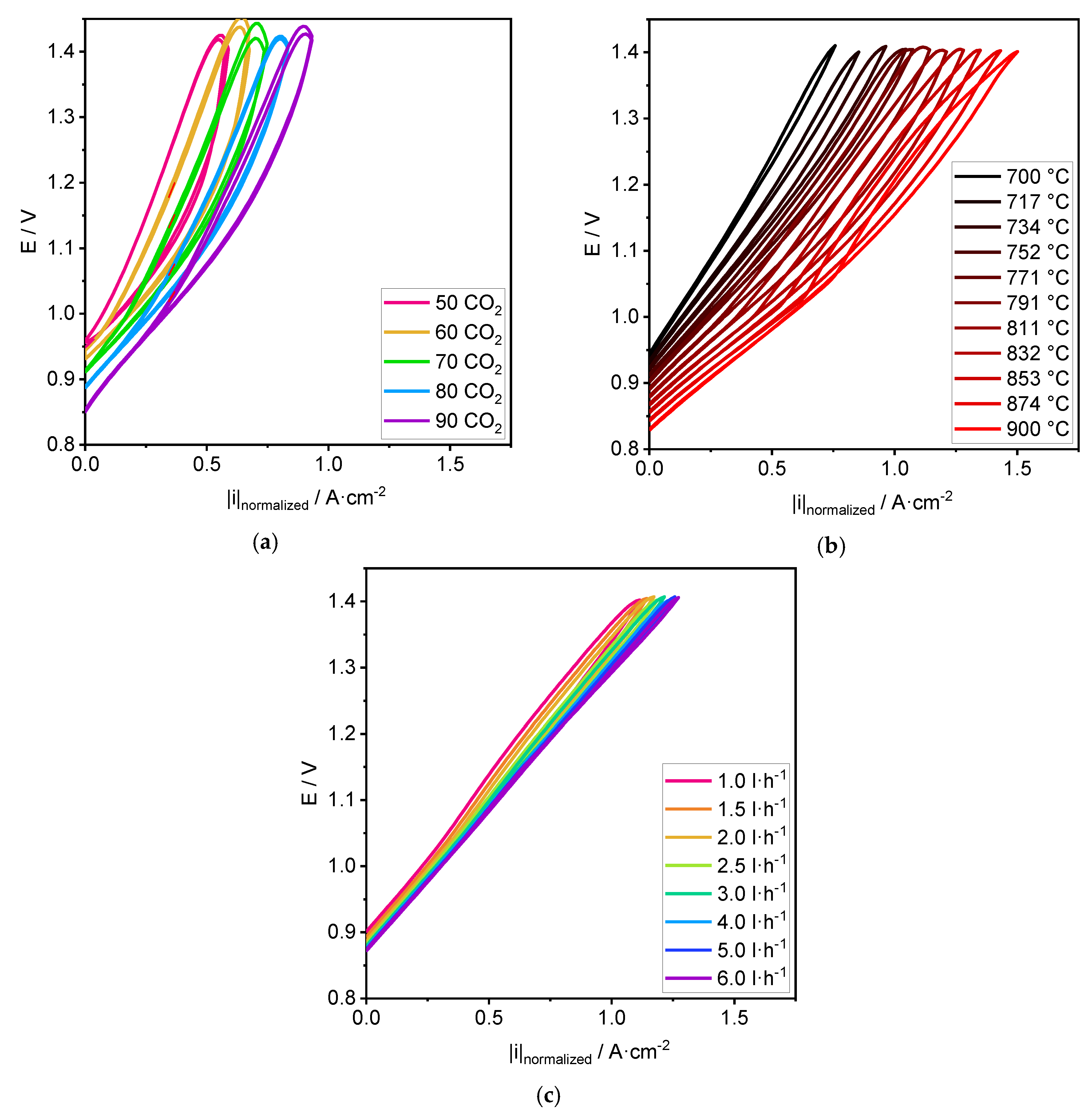
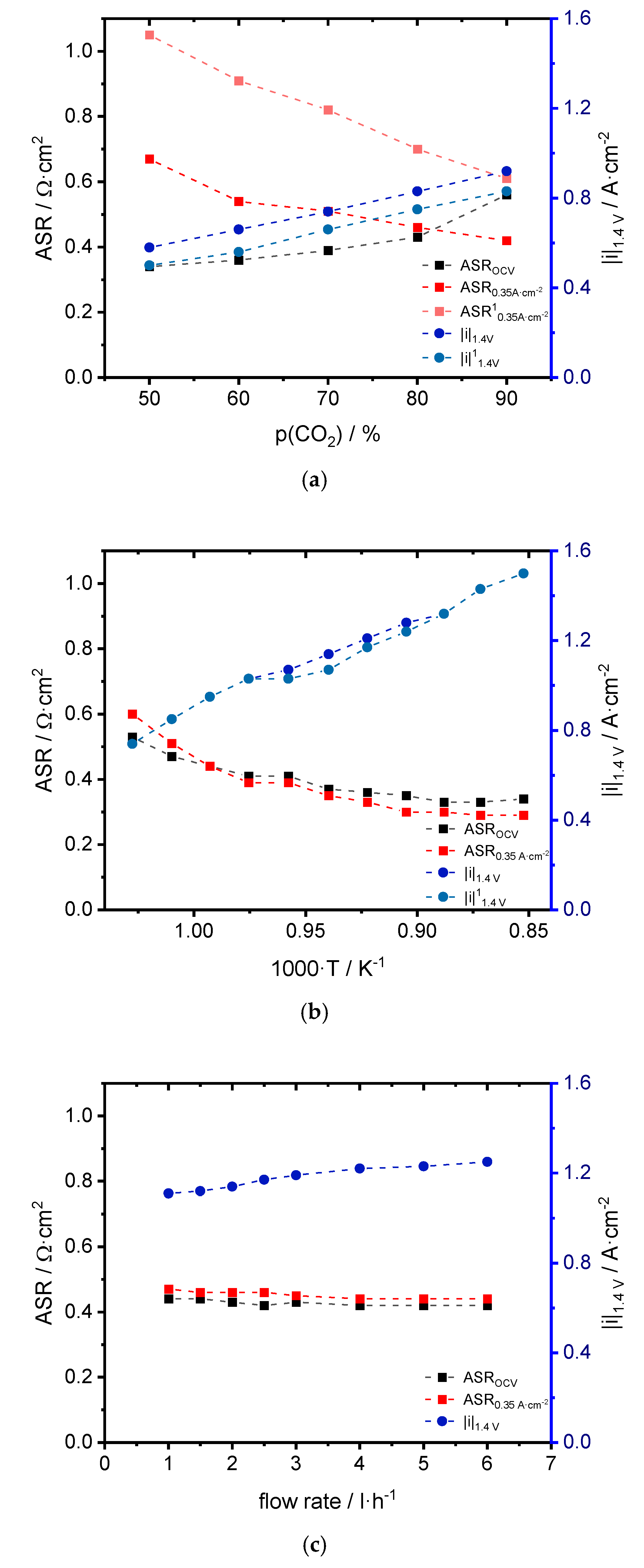
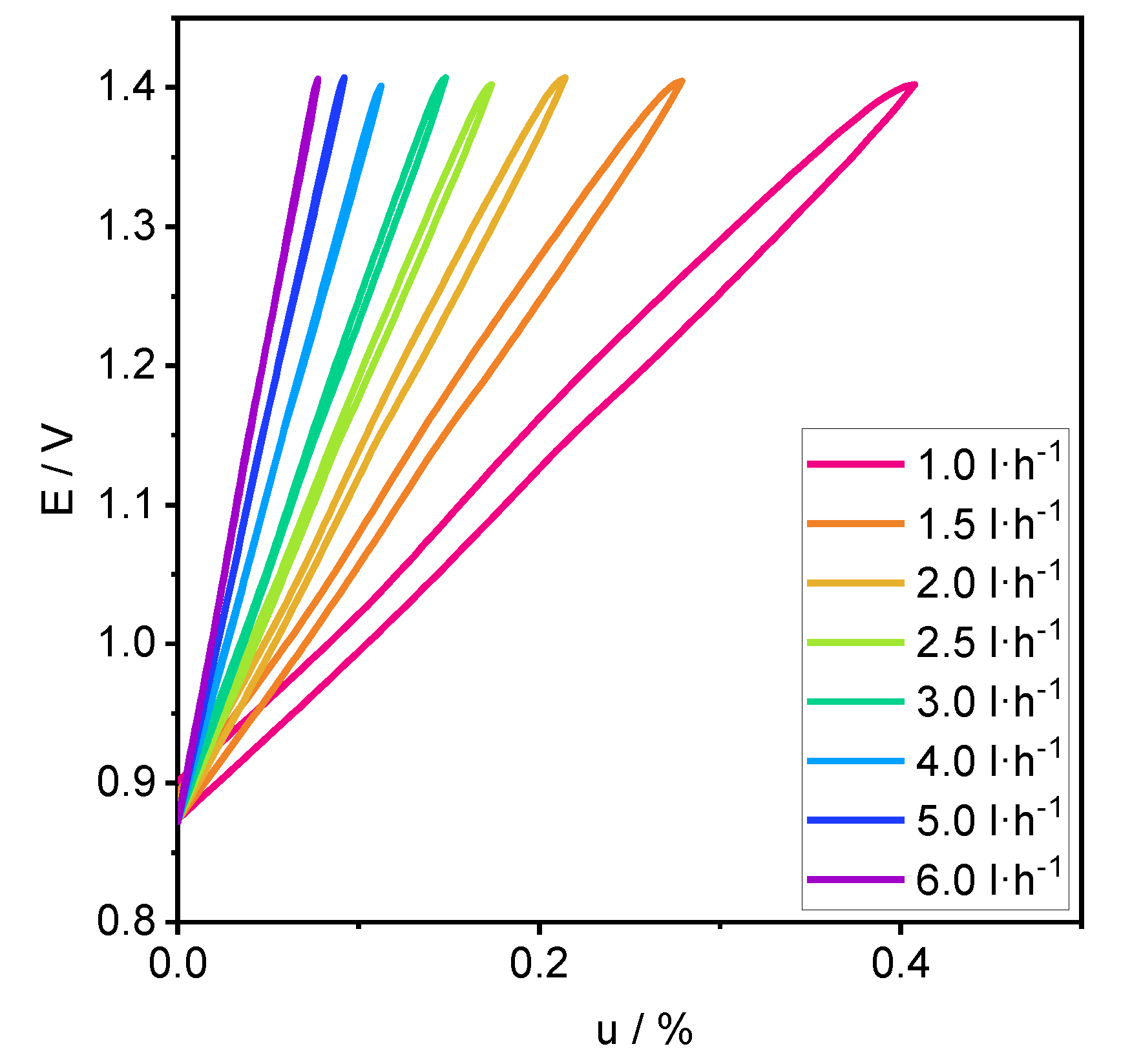
3.1. Performance
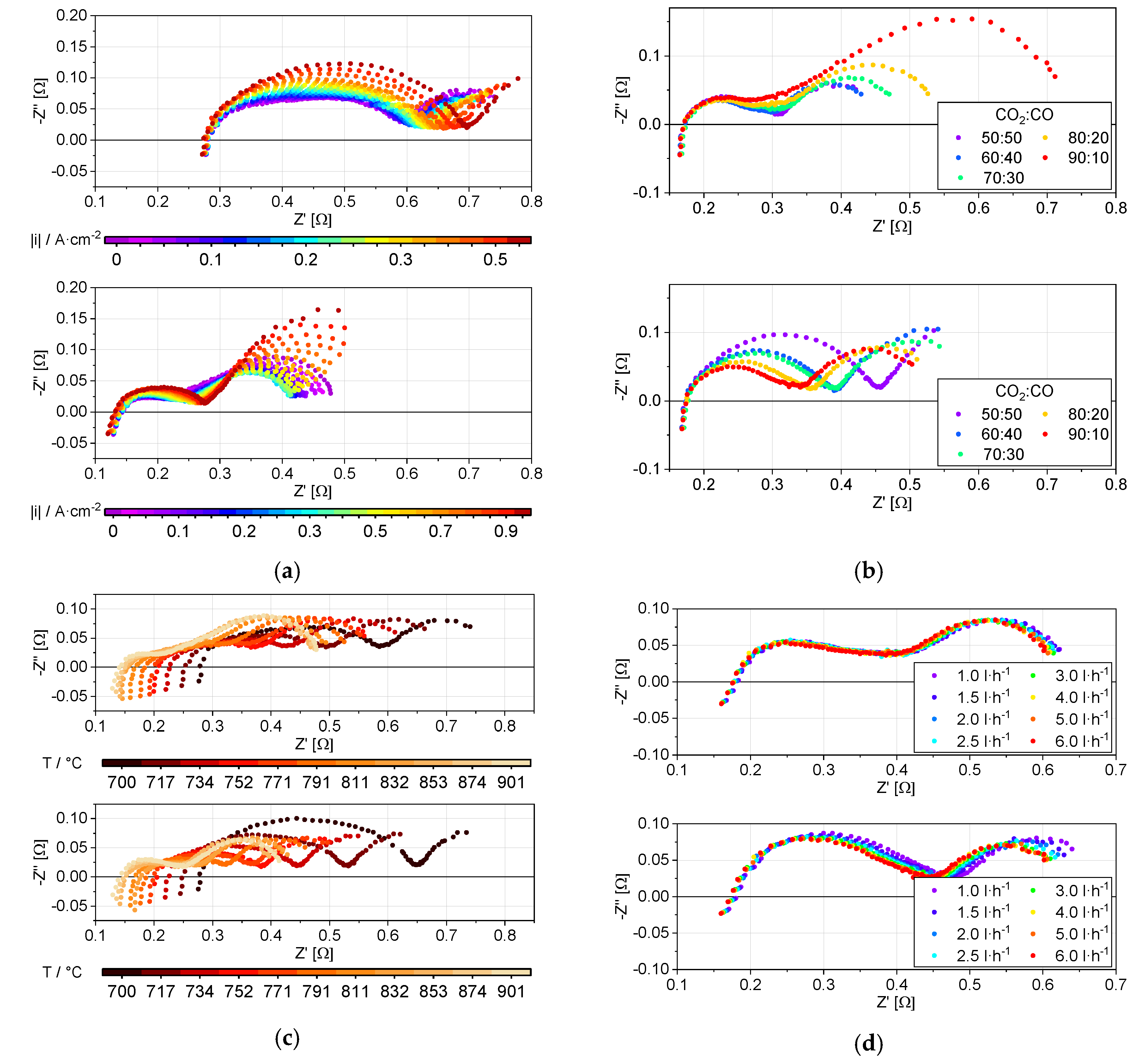
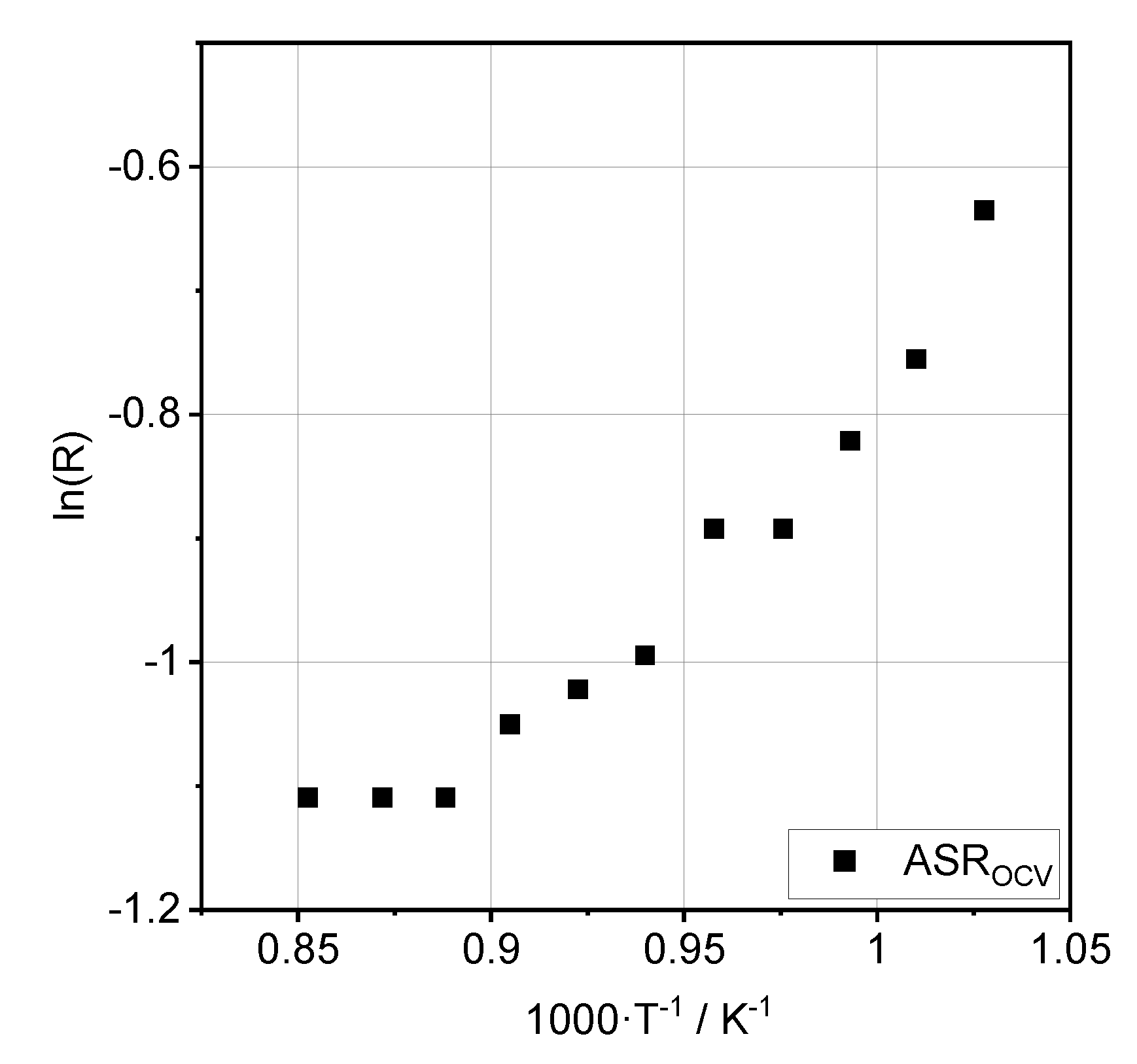
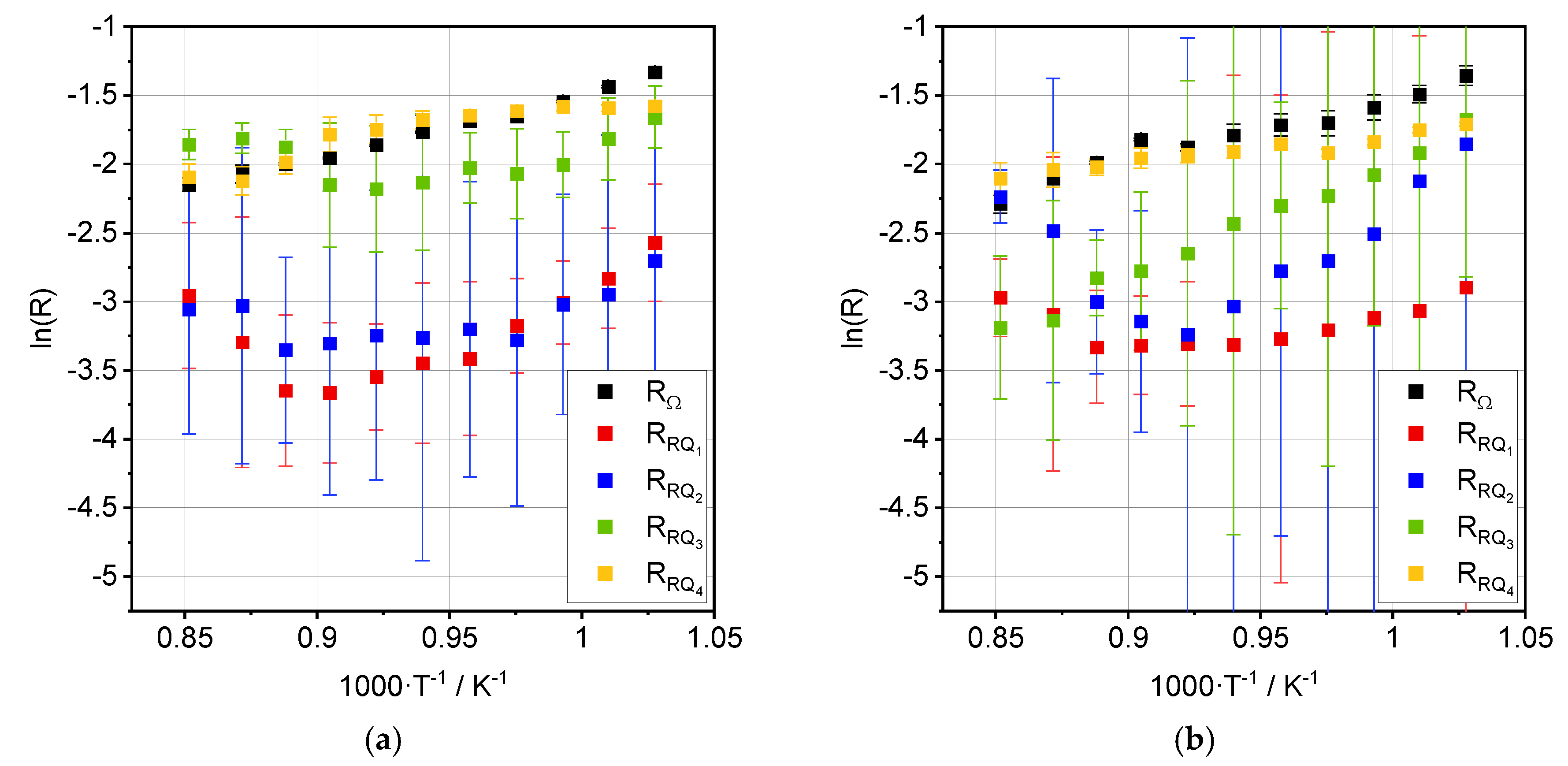
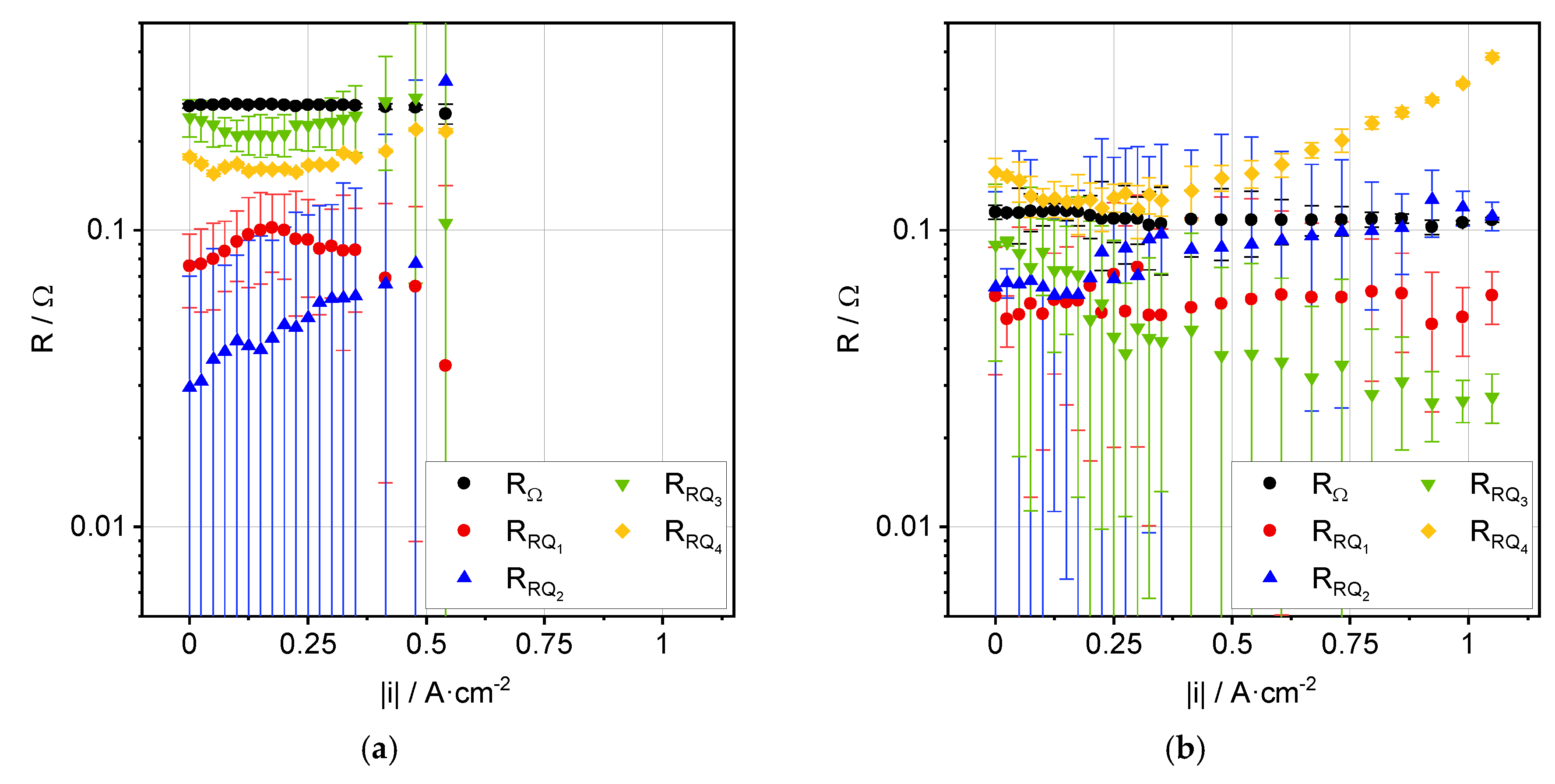
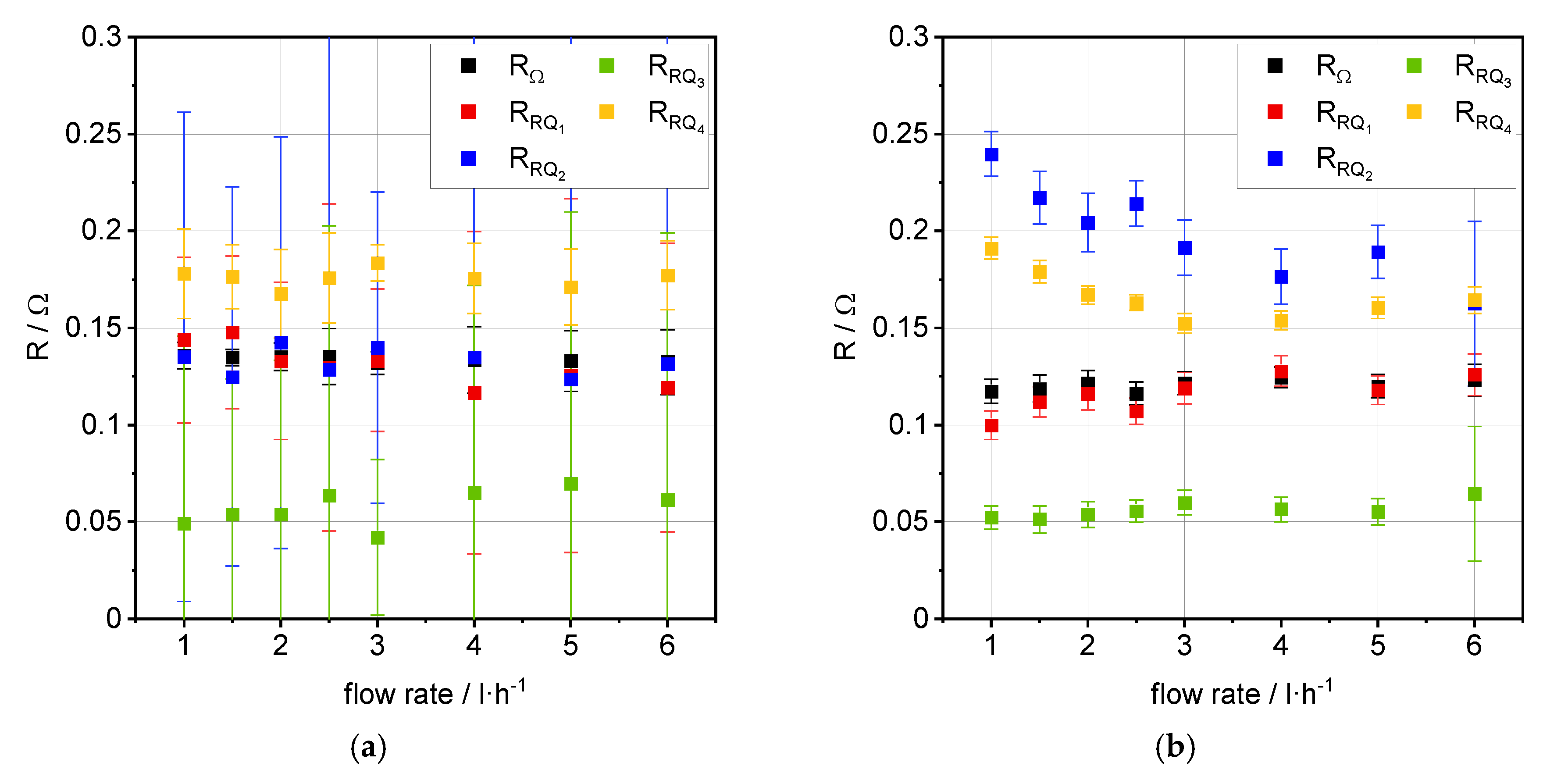
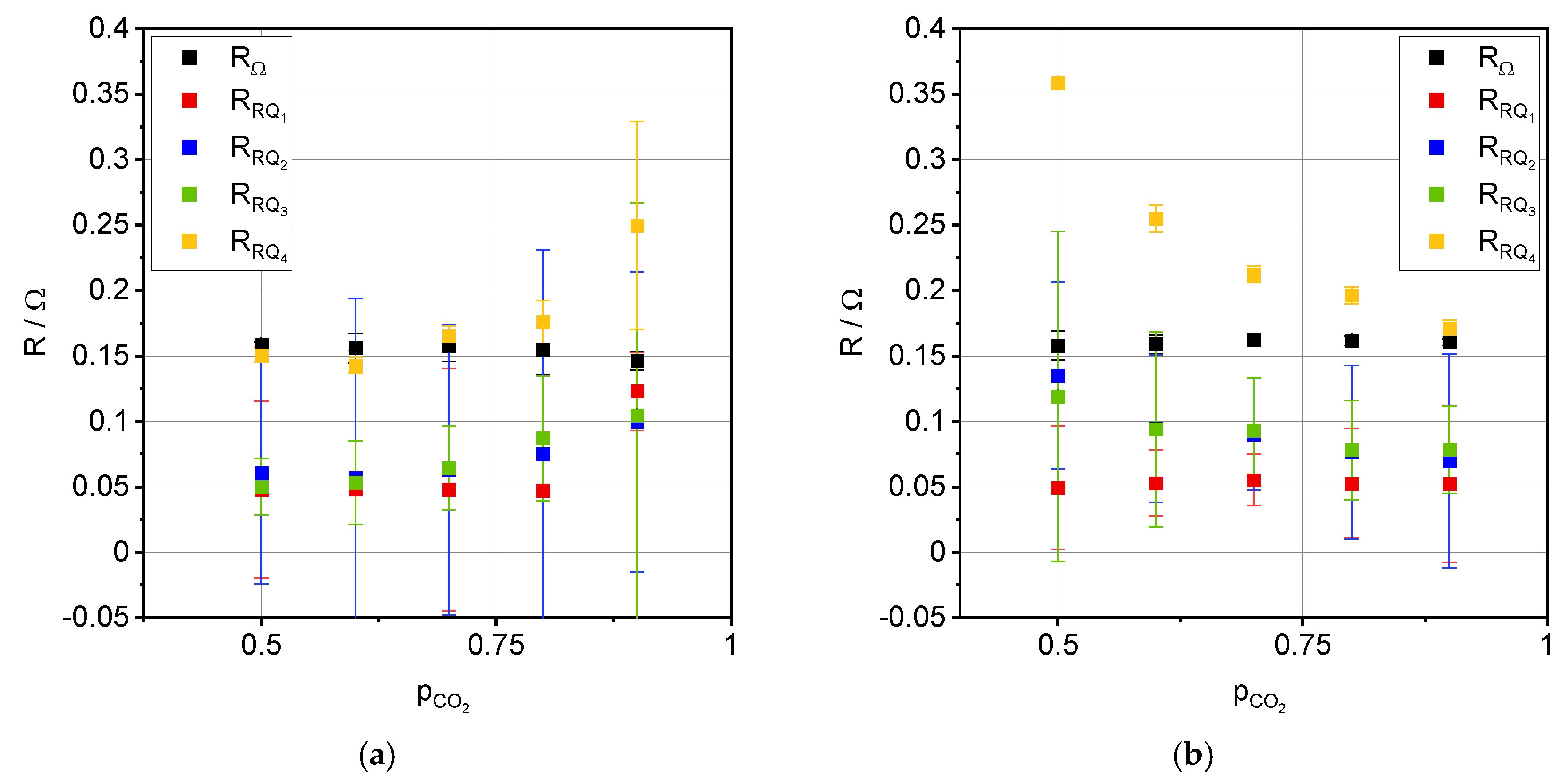
3.2. Impedance Analysis
4. Discussion
4.1. Performance
4.2. Process Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO2:CO | ASROCV [Ω·cm2] | ASR0.35 A·cm−2 [Ω·cm2] | |i|1.4 V [A·cm−2] | OCVexp [V] | OCVtheo [V] |
|---|---|---|---|---|---|
| 50:50 | 0.34 | 0.67/1.05 1 | 0.58/0.50 1 | 0.95 | 0.94 |
| 60:40 | 0.36 | 0.54/0.91 1 | 0.66/0.56 1 | 0.93 | 0.93 |
| 70:30 | 0.39 | 0.51/0.82 1 | 0.74/0.66 1 | 0.91 | 0.91 |
| 80:20 | 0.43 | 0.46/0.70 1 | 0.83/0.75 1 | 0.89 | 0.88 |
| 90:10 | 0.56 | 0.42/0.61 1 | 0.92/0.83 1 | 0.85 | 0.84 |
| T [°C] | ASROCV [Ω·cm2] | ASR0.35 A·cm−2 [Ω·cm2] | |i|1.4 V [A·cm−2] | OCVexp [V] | OCVtheo [V] |
|---|---|---|---|---|---|
| 700 | 0.53 | 0.60 | 0.74 | 0.94 | 0.94 |
| 717 | 0.47 | 0.51 | 0.85 | 0.93 | 0.93 |
| 734 | 0.44 | 0.44 | 0.95 | 0.92 | 0.92 |
| 752 | 0.41 | 0.39 | 1.03 | 0.91 | 0.91 |
| 771 | 0.41 | 0.39 | 1.07/1.03 1 | 0.90 | 0.90 |
| 791 | 0.37 | 0.35 | 1.14/1.07 1 | 0.89 | 0.89 |
| 811 | 0.36 | 0.33 | 1.21/1.17 1 | 0.88 | 0.88 |
| 832 | 0.35 | 0.30 | 1.28/1.24 1 | 0.87 | 0.86 |
| 853 | 0.33 | 0.30 | 1.32 | 0.86 | 0.85 |
| 874 | 0.33 | 0.29 | 1.43 | 0.84 | 0.84 |
| 900 | 0.34 | 0.29 | 1.50 | 0.83 | 0.83 |
| Flow Rate [L·h−1] | ASROCV [Ω·cm2] | ASR0.35 A·cm−2 [Ω·cm2] | |i|1.4 V [A·cm−2] | OCVexp [V] | OCVtheo [V] |
|---|---|---|---|---|---|
| 1.0 | 0.44 | 0.47 | 1.11 | 0.87/0.90 1 | 0.88 |
| 1.5 | 0.44 | 0.46 | 1.12 | 0.87/0.89 1 | 0.88 |
| 2.0 | 0.43 | 0.46 | 1.14 | 0.87/0.89 1 | 0.88 |
| 2.5 | 0.42 | 0.46 | 1.17 | 0.87/0.88 1 | 0.88 |
| 3.0 | 0.43 | 0.45 | 1.19 | 0.87/0.88 1 | 0.88 |
| 4.0 | 0.42 | 0.44 | 1.22 | 0.87/0.87 1 | 0.88 |
| 5.0 | 0.42 | 0.44 | 1.23 | 0.87/0.87 1 | 0.88 |
| 6.0 | 0.42 | 0.44 | 1.25 | 0.87/0.87 1 | 0.88 |
References
- Foit, S.R.; Dittrich, L.; Theuer, T.; Morgenthaler, S.; Eichel, R.-A.; de Haart, L.G.J. White Syngas by Co-Electrolysis for Industrial Chemistry. ECS Trans. 2019, 91, 2467–2474. [Google Scholar] [CrossRef]
- Dittrich, L.; Nohl, M.; Jaekel, E.E.; Foit, S.; de Haart, L.G.J.; Eichel, R.-A. High-Temperature Co-Electrolysis: A Versatile Method to Sustainably Produce Tailored Syngas Compositions. J. Electrochem. Soc. 2019, 166, F971–F975. [Google Scholar] [CrossRef]
- Theuer, T.; Schäfer, D.; Dittrich, L.; Nohl, M.; Foit, S.; Blum, L.; Eichel, R.-A.; de Haart, L.G.J. Sustainable Syngas Production by High-Temperature Co-electrolysis. Chem. Ing. Tech. 2020, 92, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Haas, T.; Krause, R.; Weber, R.; Demler, M.; Schmid, G. Technical photosynthesis involving CO2 electrolysis and fermentation. Nat. Catal. 2018, 1, 32–39. [Google Scholar] [CrossRef]
- Dittrich, L.; Nohl, M.; Theuer, T.; Foit, S.; Vinke, I.C.; de Haart, L.G.J.; Eichel, R.-A. Co-electrolysis–A sustainable technology for syngas production. Chem. Ing. Tech. 2018, 90, 1158–1159. [Google Scholar] [CrossRef] [Green Version]
- Foit, S.R.; Vinke, I.C.; de Haart, L.G.J.; Eichel, R.-A. Power-to-Syngas: An Enabling Technology for the Transition of the Energy System? Angew. Chem. Int. Ed. 2017, 56, 5402–5411. [Google Scholar] [CrossRef]
- Foit, S.R.; Dittrich, L.; Vibhu, V.; Vinke, I.C.; Eichel, R.-A.; de Haart, L.G.J. Co-Electrolysis, Quo Vadis? ECS Trans. 2017, 78, 3139–3147. [Google Scholar] [CrossRef]
- van Bavel, S.; Verma, S.; Negro, E.; Bracht, M. Integrating CO2 Electrolysis into the Gas-to-Liquids–Power-to-Liquids Process. ACS Energy Lett. 2020, 5, 2597–2601. [Google Scholar] [CrossRef]
- Küngas, R. Review—electrochemical CO2 reduction for CO production: Comparison of low- and high-temperature electrolysis technologies. J. Electrochem. Soc. 2020, 167, 044508. [Google Scholar] [CrossRef]
- Zhan, Z.; Zhao, L. Electrochemical reduction of CO2 in solid oxide electrolysis cells. J. Power Sources 2010, 195, 7250–7254. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, X.; Xie, K.; Wang, G.; Bao, X. High-temperature CO2 electrolysis in solid oxide electrolysis cells: Developments, challenges, and prospects. Adv. Mater. 2019, 31, 1902033. [Google Scholar] [CrossRef]
- Song, Y.; Zhou, Z.; Zhang, X.; Zhou, Y.; Gong, H.; Lv, H.; Liu, Q.; Wang, G.; Bao, X. Pure CO2 electrolysis over an Ni/YSZ cathode in a solid oxide electrolysis cell. J. Mater. Chem. A 2018, 6, 13661–13667. [Google Scholar] [CrossRef]
- Skafte, T.L.; Blennow, P.; Hjelm, J.; Graves, C. Carbon deposition and sulfur poisoning during CO2 electrolysis in nickel-based solid oxide cell electrodes. J. Power Sources 2018, 373, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Song, Y.; Wang, G.; Bao, X. Co-electrolysis of CO2 and H2O in high-temperature solid oxide electrolysis cells: Recent advance in cathodes. J. Energy Chem. 2017, 26, 839–853. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Chen, X.; Wu, J.; Sheng, Z.; Liu, G.; Wang, Y. A highly-performed, dual-layered cathode supported solid oxide electrolysis cell for efficient CO2 electrolysis fabricated by phase inversion co-tape casting method. J. Electrochem. Soc. 2017, 164, F1130–F1135. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Yao, W.; Dong, D.; Xie, K. Direct electrolysis of CO2 using an oxygen-ion conducting solid oxide electrolyzer based on La0.75Sr0.25Cr0.5Mn0.5O3−δ electrode. J. Power Sources 2013, 230, 115–121. [Google Scholar] [CrossRef]
- Shi, Y.; Luo, Y.; Cai, N.; Qian, J.; Wang, S.; Li, W.; Wang, H. Experimental characterization and modeling of the electrochemical reduction of CO2 in solid oxide electrolysis cells. Electrochim. Acta 2013, 88, 644–653. [Google Scholar] [CrossRef]
- Bidrawn, F.; Kim, G.; Corre, G.; Irvine, J.T.S.; Vohs, J.M.; Gorte, R.J. Efficient reduction of CO2 in a solid oxide electrolyzer. Electrochem. Solid-State Lett. 2008, 11, B167. [Google Scholar] [CrossRef]
- Udomsilp, D.; Roehrens, D.; Menzler, N.H.; Bischof, C.; de Haart, L.G.J.; Opitz, A.K.; Guillon, O.; Bram, M. High-performance metal-supported solid oxide fuel cells by advanced cathode processing. J. Electrochem. Soc. 2017, 164, F1375–F1384. [Google Scholar] [CrossRef] [Green Version]
- Leonide, A.; Sonn, V.; Weber, A.; Ivers-Tiffée, E. Evaluation and modeling of the cell resistance in anode-supported solid oxide fuel cells. J. Electrochem. Soc. 2008, 155, B36. [Google Scholar] [CrossRef]
- Korte, C.; Peters, A.; Janek, J.; Hesse, D.; Zakharov, N. Ionic conductivity and activation energy for oxygen ion transport in superlattices--the semicoherent multilayer system YSZ (ZrO2 + 9.5 mol% Y2O3)/Y2O3. Phys. Chem. Chem. Phys. 2008, 10, 4623–4635. [Google Scholar] [CrossRef]
- Badwal, S.P.S. Zirconia-based solid electrolytes: Microstructure, stability and ionic conductivity. Solid State Ionics 1992, 52, 23–32. [Google Scholar] [CrossRef]
- Li, W.; Shi, Y.; Luo, Y.; Wang, Y.; Cai, N. Carbon monoxide/carbon dioxide electrochemical conversion on patterned nickel electrodes operating in fuel cell and electrolysis cell modes. Int. J. Hydrogen Energy 2016, 41, 3762–3773. [Google Scholar] [CrossRef]
- Hecht, E.S.; Gupta, G.K.; Zhu, H.; Dean, A.M.; Kee, R.J.; Maier, L.; Deutschmann, O. Methane reforming kinetics within a Ni–YSZ SOFC anode support. Appl. Catal. A Gen. 2005, 295, 40–51. [Google Scholar] [CrossRef]
- Sonn, V.; Leonide, A.; Ivers-Tiffée, E. Combined deconvolution and CNLS fitting approach applied on the impedance response of technical Ni∕8YSZ cermet electrodes. J. Electrochem. Soc. 2008, 155, B675. [Google Scholar] [CrossRef]
- Primdahl, S.; Mogensen, M. Gas diffusion impedance in characterization of solid oxide fuel cell anodes. J. Electrochem. Soc. 1999, 146, 2827–2833. [Google Scholar] [CrossRef]
- Ebbesen, S.D.; Sun, X.; Mogensen, M.B. Understanding the processes governing performance and durability of solid oxide electrolysis cells. Faraday Discuss. 2015, 182, 393–422. [Google Scholar] [CrossRef] [PubMed]









| Process | Ea, OCV [kJ·mol−1] | Ea, 0.35 A·cm−2 [kJ·mol−1] |
|---|---|---|
| RΩ | 40.6 ± 1.5 | 38.1 ± 2.0 |
| RRQ1 | 71.8 ± 5.4 | 18.5 ± 3.8 |
| RRQ2 | 36.2 ± 8.0 | 80.2 ± 8.4 |
| RRQ3 | 36.3 ± 7.3 | 71.8 ± 2.4 |
| RRQ4 | 10.0 ± 2.1 | 19.1 ± 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foit, S.; Dittrich, L.; Duyster, T.; Vinke, I.; Eichel, R.-A.; de Haart, L.G.J. Direct Solid Oxide Electrolysis of Carbon Dioxide: Analysis of Performance and Processes. Processes 2020, 8, 1390. https://doi.org/10.3390/pr8111390
Foit S, Dittrich L, Duyster T, Vinke I, Eichel R-A, de Haart LGJ. Direct Solid Oxide Electrolysis of Carbon Dioxide: Analysis of Performance and Processes. Processes. 2020; 8(11):1390. https://doi.org/10.3390/pr8111390
Chicago/Turabian StyleFoit, Severin, Lucy Dittrich, Tobias Duyster, Izaak Vinke, Rüdiger-A. Eichel, and L. G. J. (Bert) de Haart. 2020. "Direct Solid Oxide Electrolysis of Carbon Dioxide: Analysis of Performance and Processes" Processes 8, no. 11: 1390. https://doi.org/10.3390/pr8111390
APA StyleFoit, S., Dittrich, L., Duyster, T., Vinke, I., Eichel, R. -A., & de Haart, L. G. J. (2020). Direct Solid Oxide Electrolysis of Carbon Dioxide: Analysis of Performance and Processes. Processes, 8(11), 1390. https://doi.org/10.3390/pr8111390