1. Introduction
The research exploration of new inorganic materials with open framework constructed of polyhedra sharing faces, edges and/or corners forming 1D channels, 2D inter-layer spaces or 3D networks where cations are located, is currently an area of intense activity including several disciplines, in particular solid-state chemistry. In particular, alkali metal phosphates were found to have various applications because of their electric, piezoelectric, ferroelectric, magnetic, and catalytic properties [
1,
2,
3,
4]. Among those, the families of materials with the melilite structure [
5], the olivine structure [
6] and the natrium super ionic conductor (NaSICON) structure [
7], attracted attention for their ionic conduction and exchange of ions [
6,
7].
More recently, in a series of studies arsenate analogs have been synthesized [
8,
9,
10]. But, until today phosphate compounds are more studied as cathodes [
11,
12] compared to arsenate and this is perhaps due to the toxicity of arsenic III (As
2O
3). However, the oxide of arsenic V (As
2O
5) is less toxic. In addition, the introduction of arsenic into a structure changes its physical and chemical properties and even toxicity. On the other hand, the comparison of the electrochemical properties of LiCoPO
4 and LiCoAsO
4 both with olivine structure and close unit cell parameters, shows reversible (de)intercalation from/into material at average voltages of 4.8 and 4.6 V, respectively for LiCoAsO
4 [
10] and a voltage average of 2.5–5 V for LiCoPO
4 [
11].
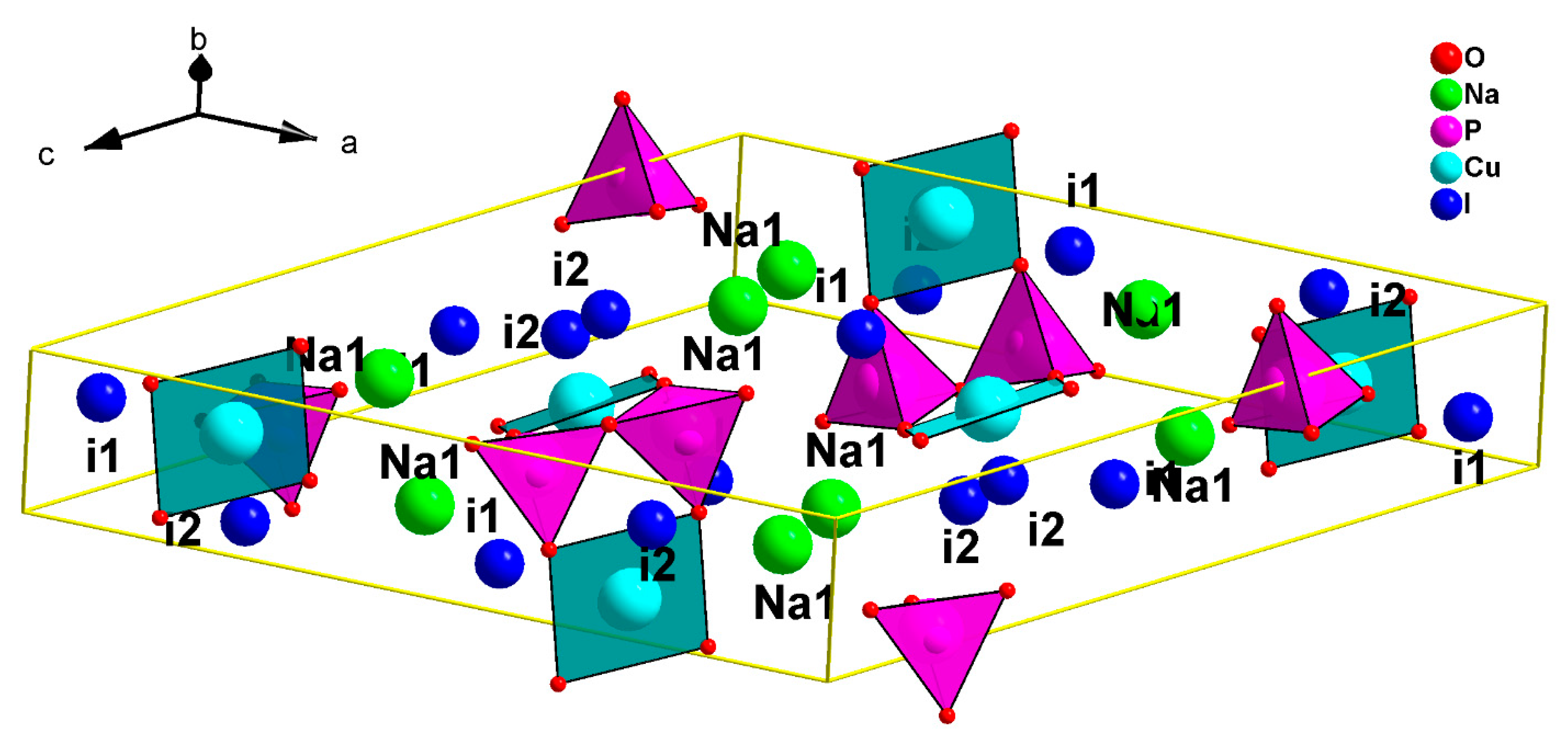
The Na
2MP
2O
7 systems (
M = transition metal) [
13,
14] have a layered structure with the layers [
MP
2O
7]
n2n− and the sodium cations localized in the interlayer space, which favors high ionic conductivity. We recently investigated the effect of the substitution of P by As with a larger ionic radius for ionic conductivity and showed the improvement of ionic conductivity for Na
2CoP
1.5As
0.5O
7 [
15], which has an electrical conductivity value of σ
240 °C = 7.91×10
−5 Scm
−1 and an activation energy Ea = 0.56 eV compared to Na
2CoP
2O
7 (σ
300 °C = 2 × 10
−5 Scm
−1; Ea = 0.63 eV) [
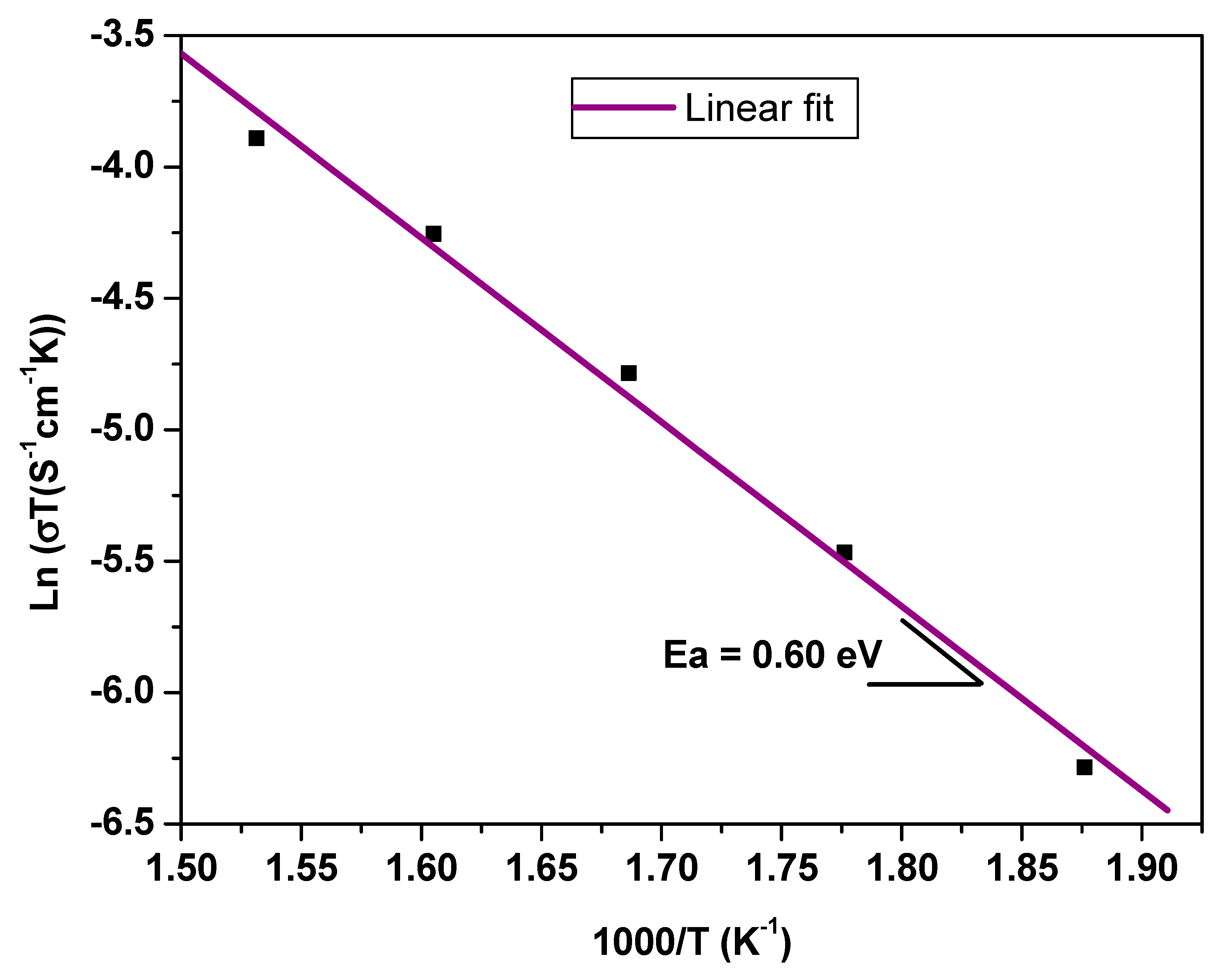
13].
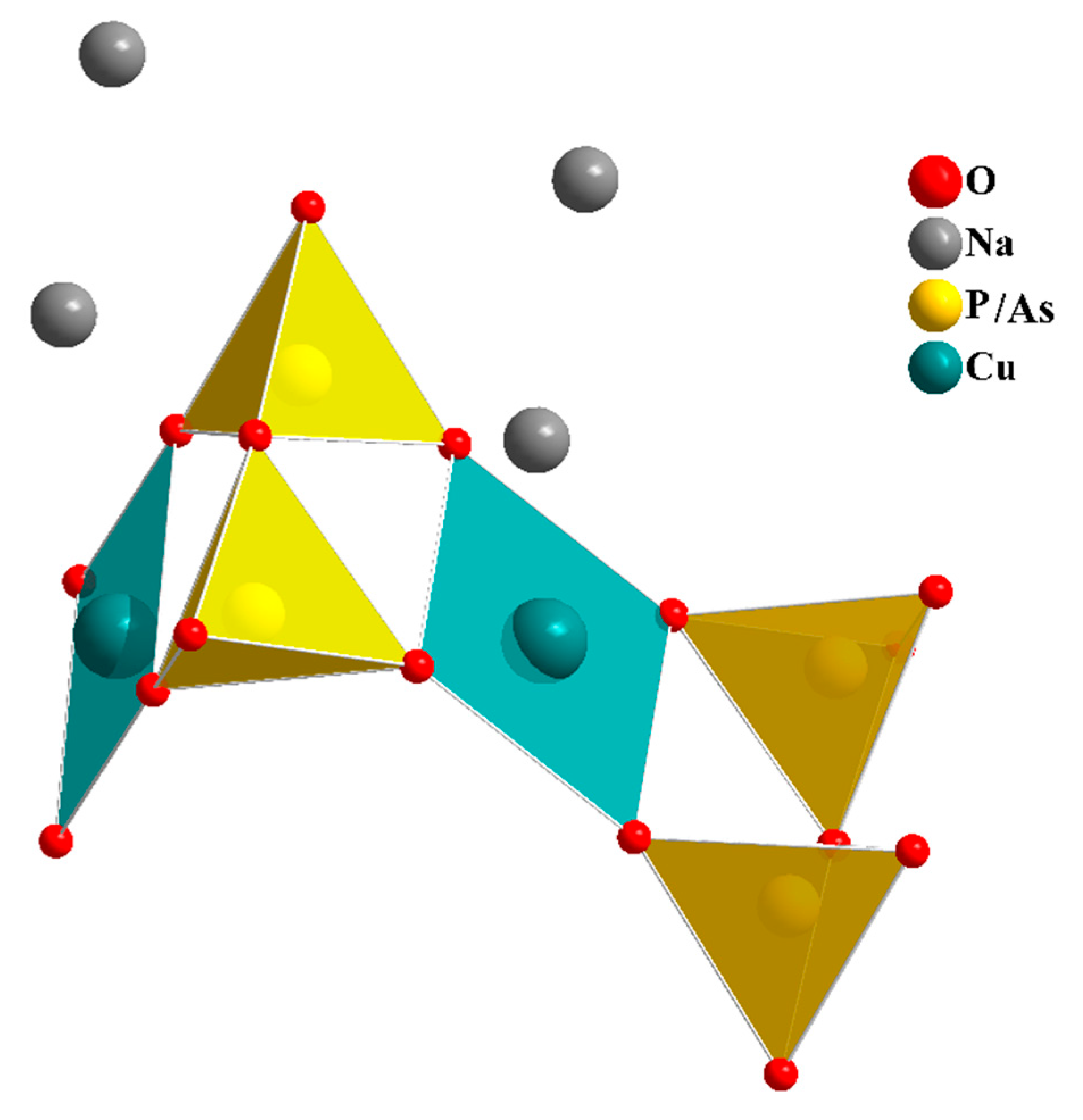
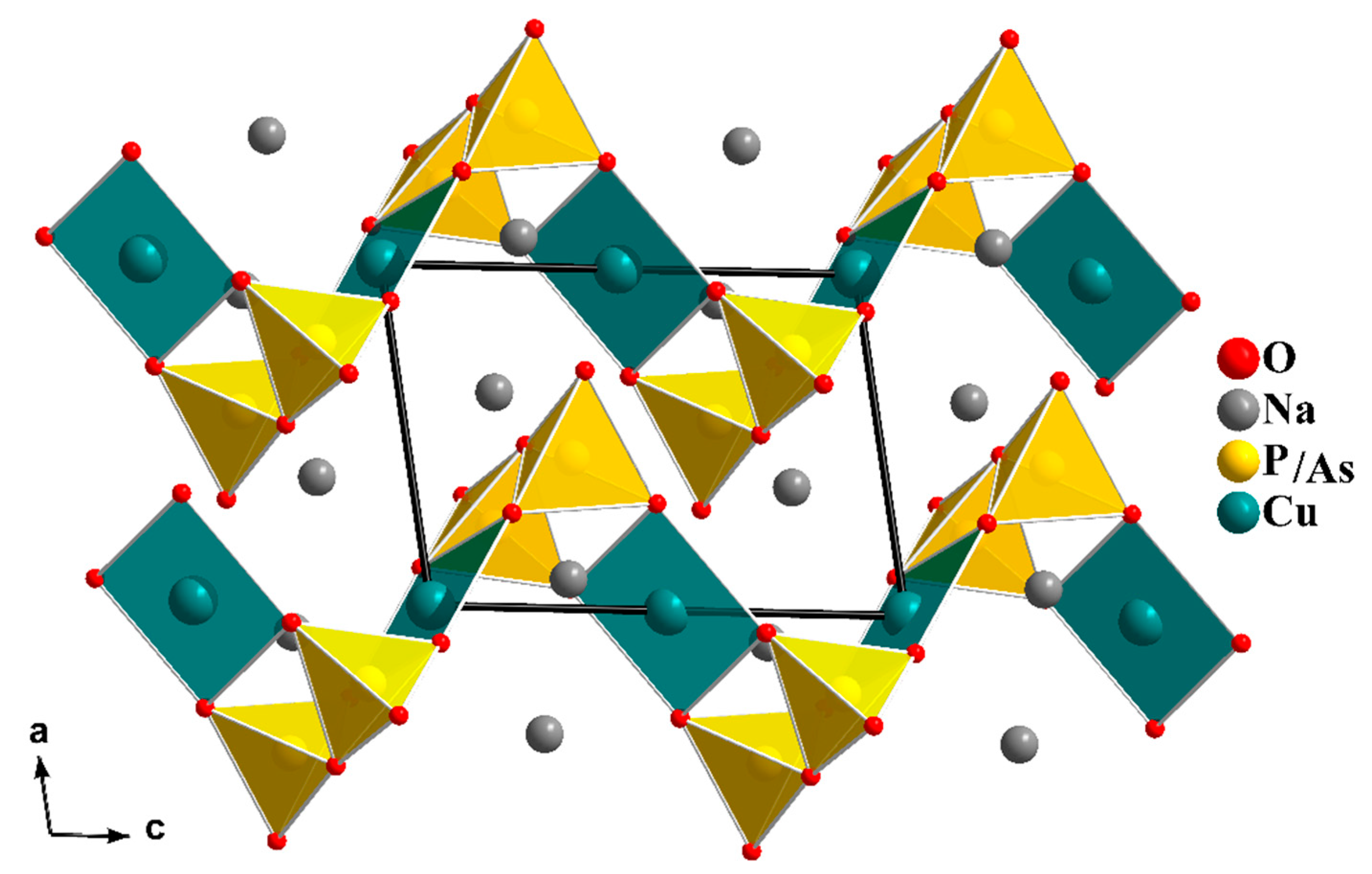
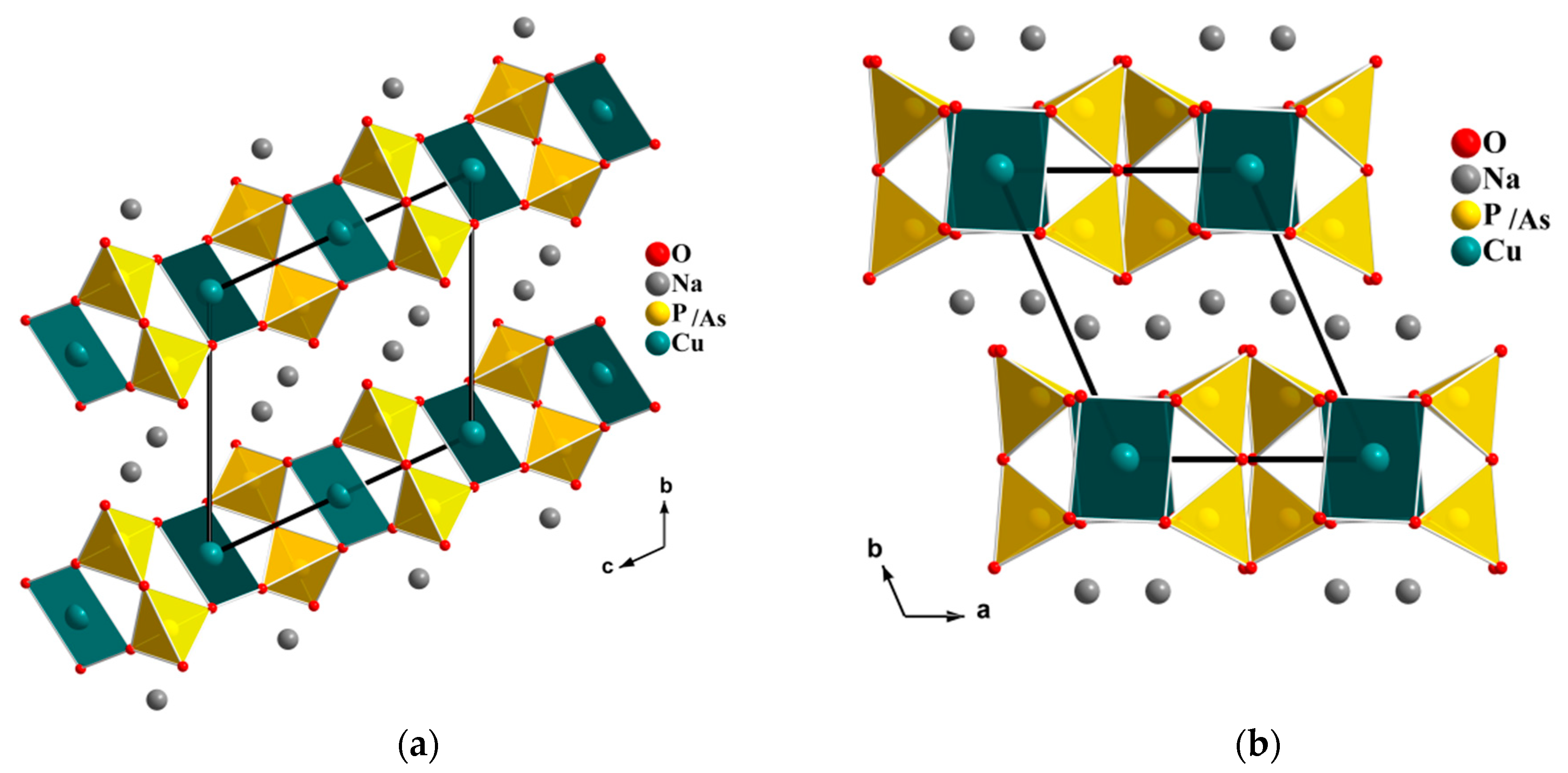
In our search for new polyanion oxides of sodium and transition metals, the exploration of the Na2O-CuO-P2O5-As2O5 crystallographic systems allowed us to isolate a new member of di-phosphate arsenates, Na2CuP1.5As0.5O7, in the polycrystalline powder form. In this paper, characterizations and physicochemical studies of the new member of sodium copper diphosphate-diarsenate material, Na2CuP1.5As0.5O7, and a comparison with other previous works encountered in the literature will be presented.
2. Materials and Methods
A mixture of Cu(NO3)2.2.5H2O, NH4H2PO4 and Na2HAsO4.7H2O in the molar ratio Na:Cu:P:As equal to 2:1:1.5:0.5 was placed in a porcelain crucible and heated to 350 °C for 24 h to eliminate the volatile products H2O, NO2, and NH3. The obtained powder was ground manually using agate mortar and shaped as cylindrical pellets by a uniaxial press. The obtained pellets were heated to 600 °C. After 72 h, the sample was cooled slowly at a rate of 10 °C/h down to room temperature. After grinding finely, a blue polycrystalline powder was obtained.
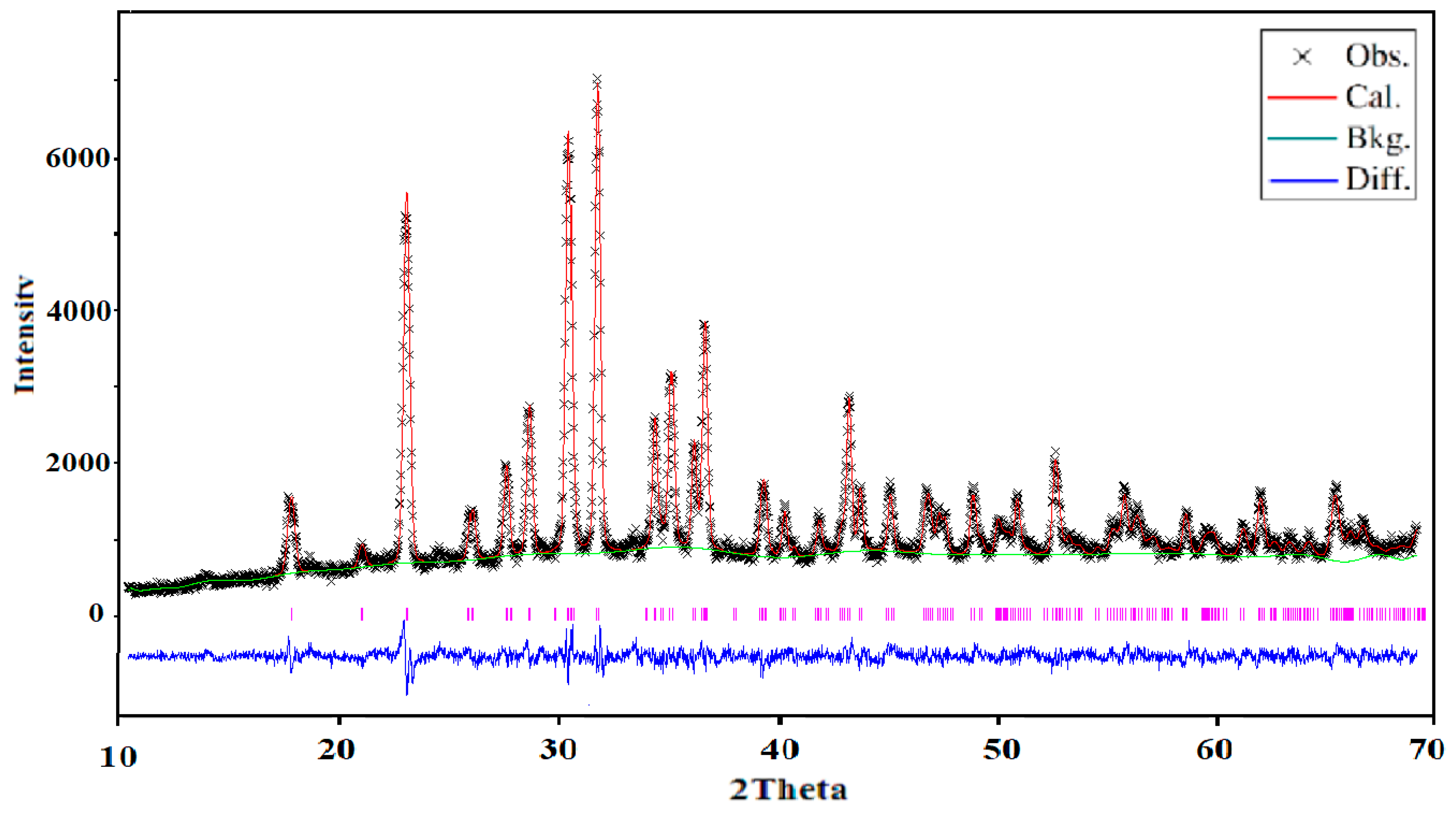
X-ray powder diffraction (XRD) was used to control and ensure the purity of the obtained powder. The analysis was carried out using XRD-6000 (Shimadzu, Japan) with graphite monochromator (Cukα, λ = 0.154178 nm) and a scan range of 2θ = 10°–70° with step of about 0.02°. The structure was refined using the Rietveld method by the means of the GSAS computer program [
16] (EXPGUI, Gaithersburg, Maryland, USA). The crystallographic data of Na
2CuP
2O
7 [
17] was used as a starting set. The obtained structural model was confirmed by the Charge Distribution CHARDI model of validation. The CHARDI calculation was done by using the CHARDI2015 computer program (Nespolo, IUCR) [
18].
FTIR spectrometer (Agilent Technologies Cary 630 model) was used to allow a direct indexation of the peaks on a spectral range in wave numbers ranging from (1300–400 cm−1).
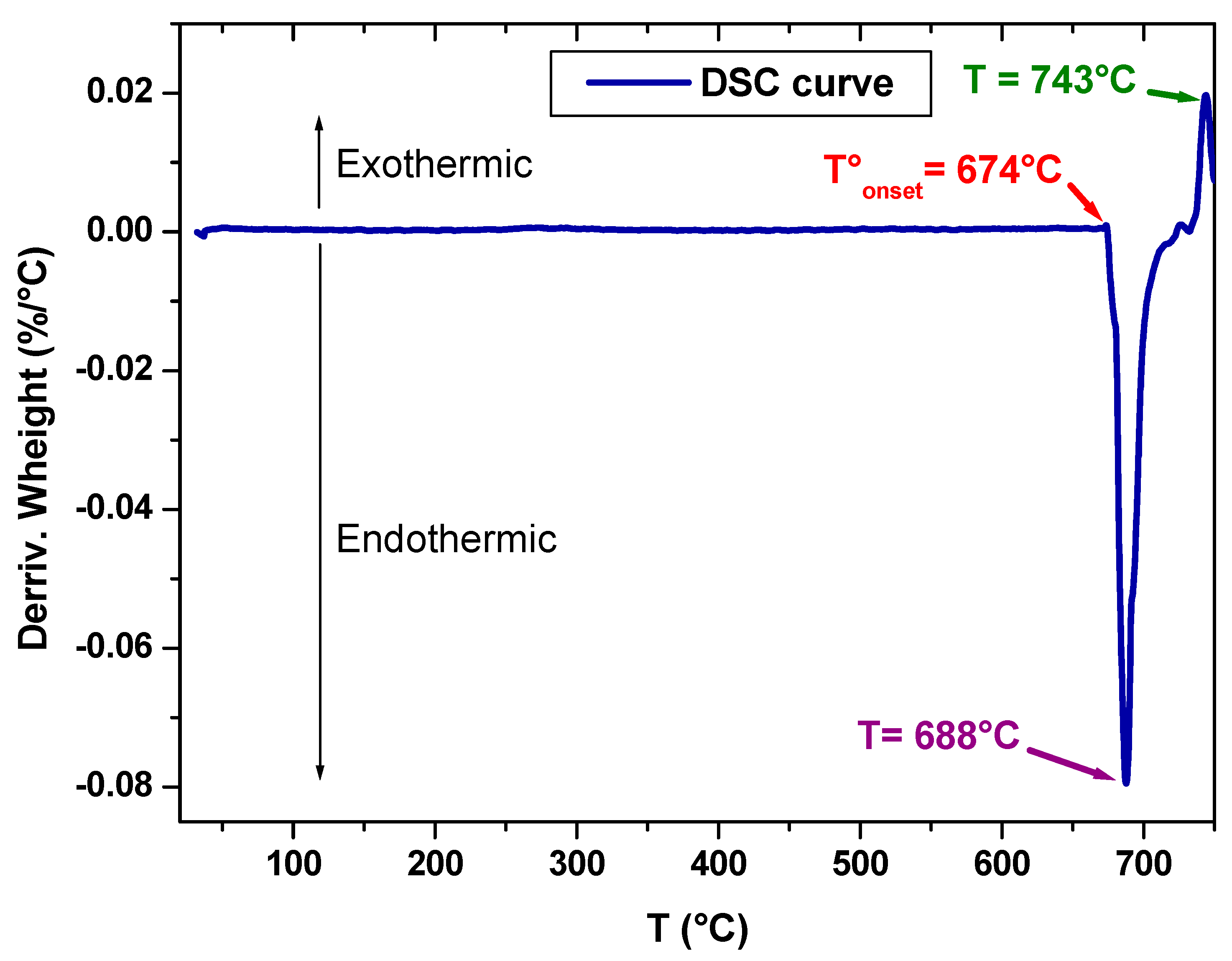
Differential scanning calorimetry (DSC), with the SDT Q600 model, was used to study the thermal behavior of the obtained and prepared sample. In fact, the device contains two crucibles, one as a reference and the other contains the sample to be analyzed. These two crucibles are heated to 750 °C at a rate of 10 °C/min. The thermal analysis was carried out under a nitrogen atmosphere to avoid the reaction of the sample with the oxygen in the air.
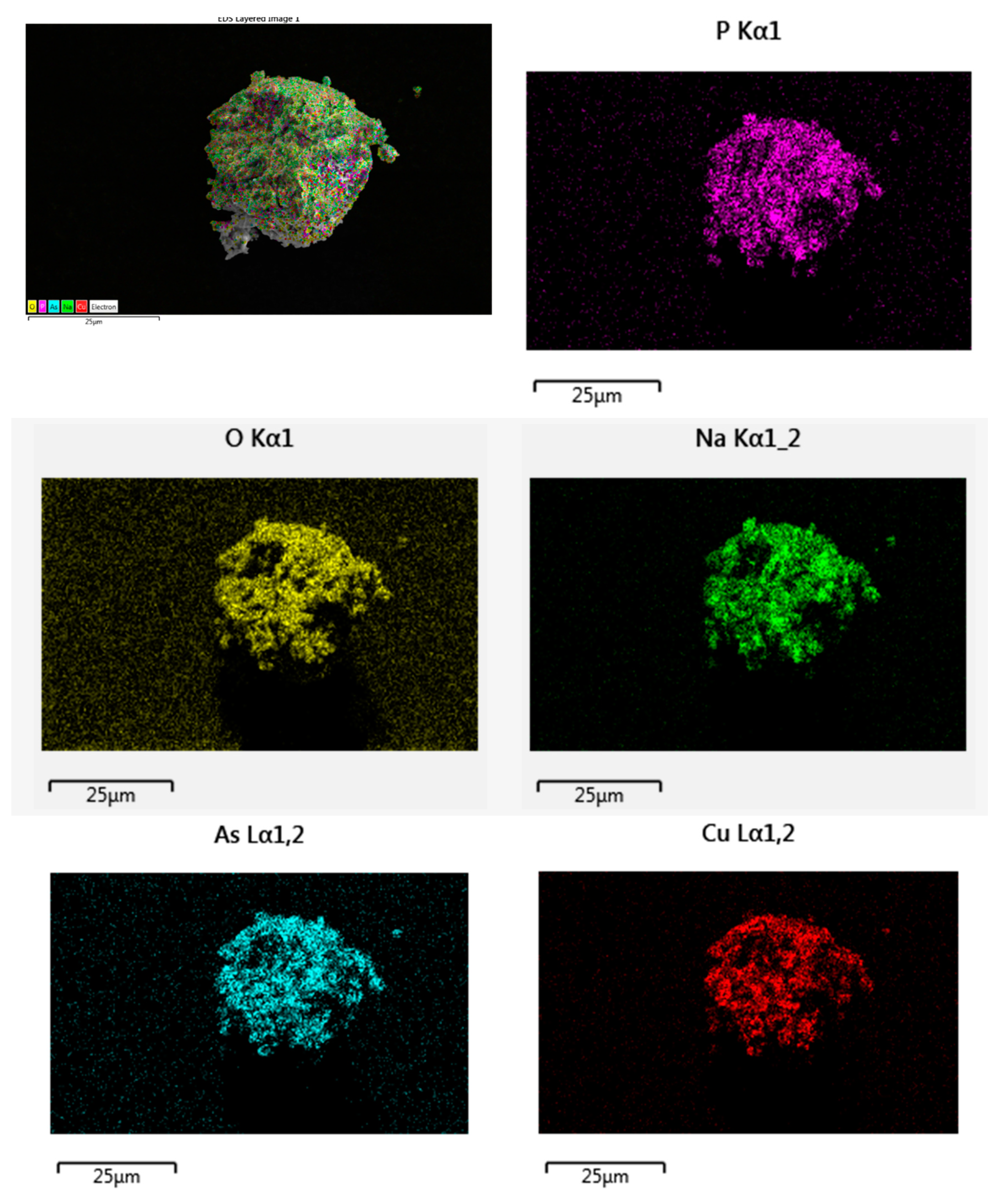
Energy-dispersive X-ray spectroscopy (EDX) and scanning electron microscopy (SEM, Thermo Fisher Scientific model), were used to identify the present elements and the microstructure of the studied material, respectively.
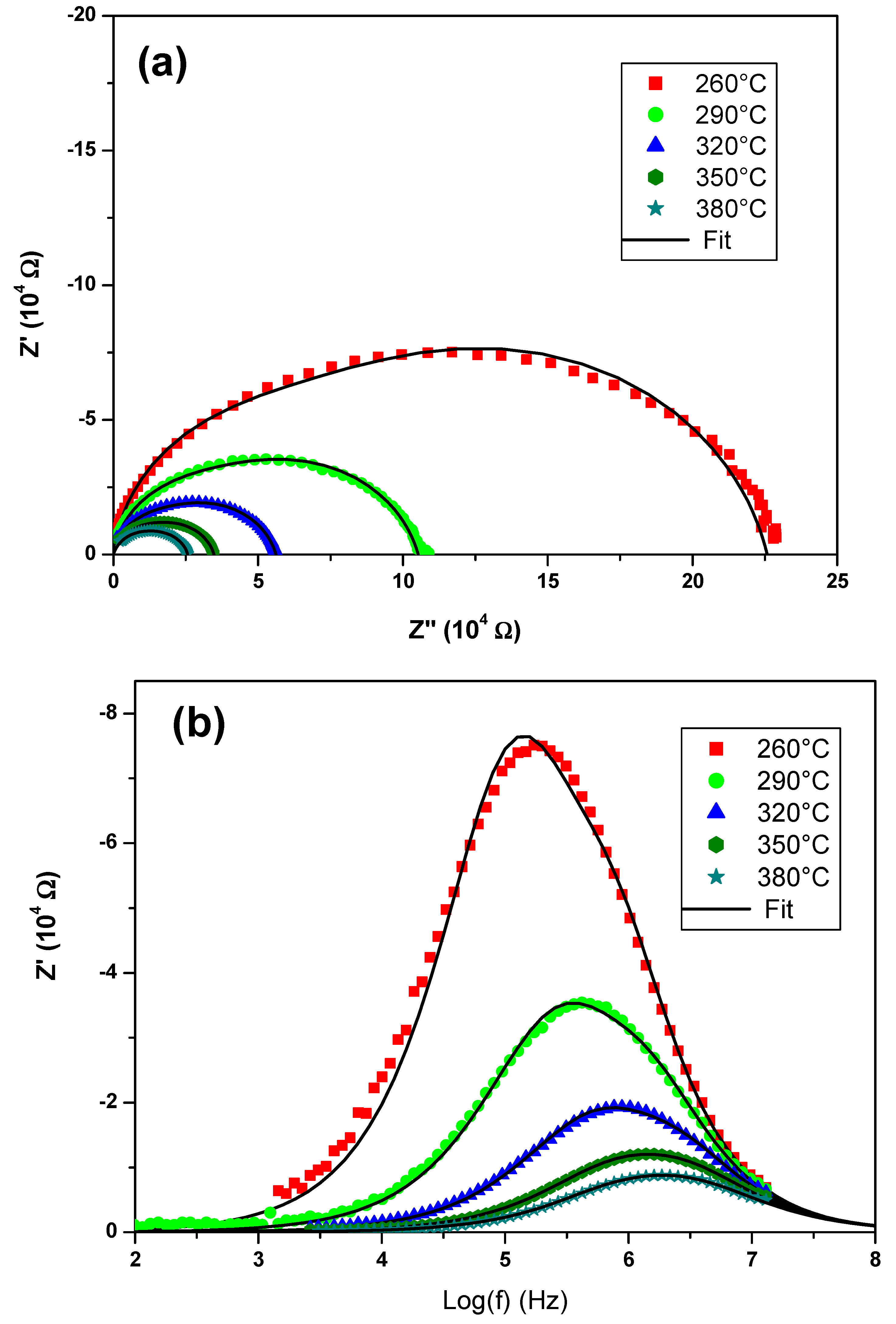
The electrical measurements were preceded by pretreatment of the sample in order to densify the measured sample by reducing the mean particle size of the synthesized powder. Mechanical grinding for 100 min was carried out using the FRISCH planetary micromill pulverisette 7. The polycrystalline sample was shaped as a cylindrical pellet using a uniaxial press. The pellet was sintered in air at an optimal temperature of 610 °C for 2 h with 5 °C/min heating and cooling rates. The geometric factor of the dense ceramic is g = e/S = 0.793 cm−1 where e and S are the thickness and face area of pellet, respectively. Gold metal electrodes ~36 nm thick were deposited using a SC7620 mini sputter coater. The sample was then placed between two platinum electrodes that were connected by platinum cables to the frequency response analyzer (HP 4192A) which was controlled by a microcomputer.
Impedance spectroscopic measurements were performed via the Hewlett-Packard 4192-A automatic bridge supervised by HP workstation. Impedance spectra were recorded with 0.5 V AC-signal in the 5 Hz–13 MHz frequency range.
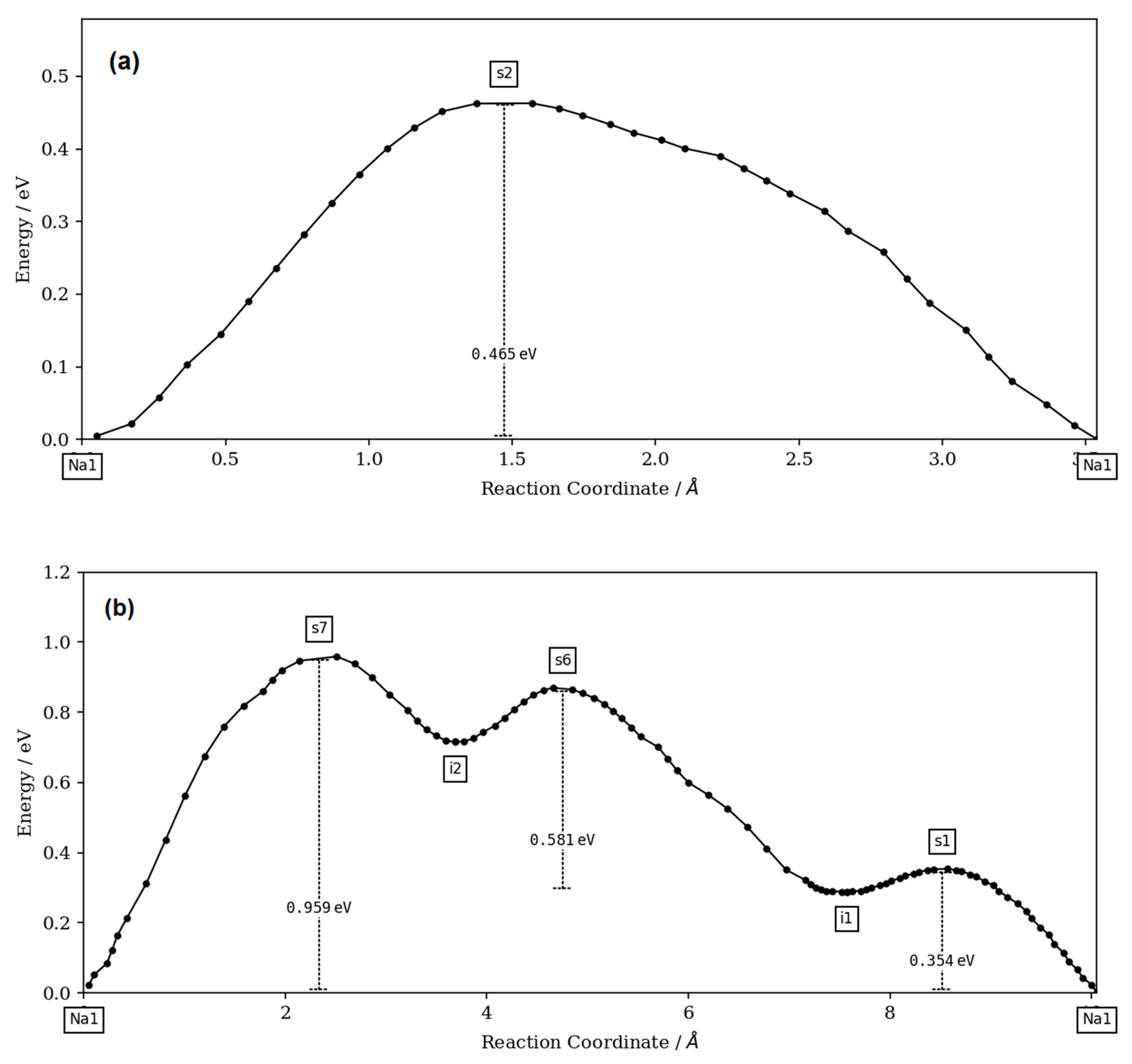
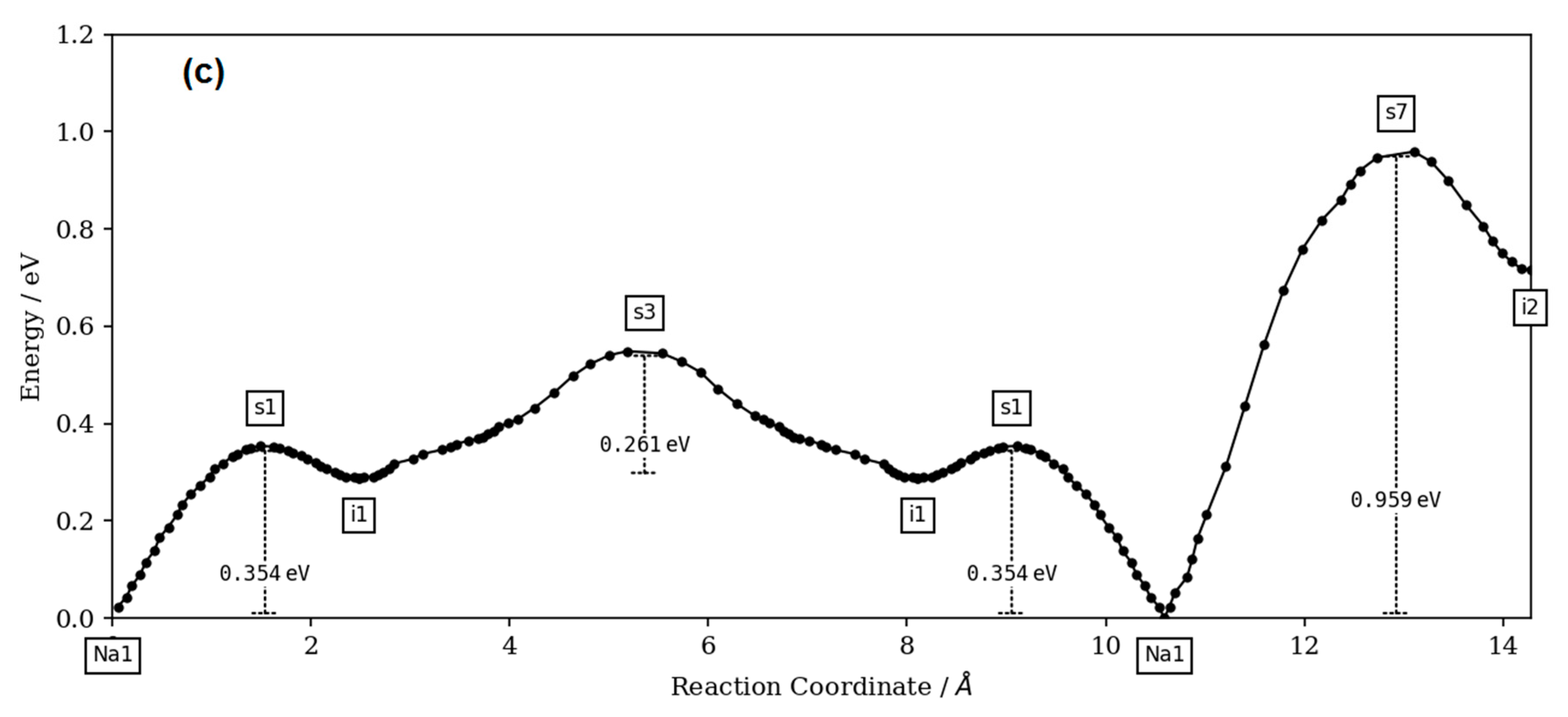
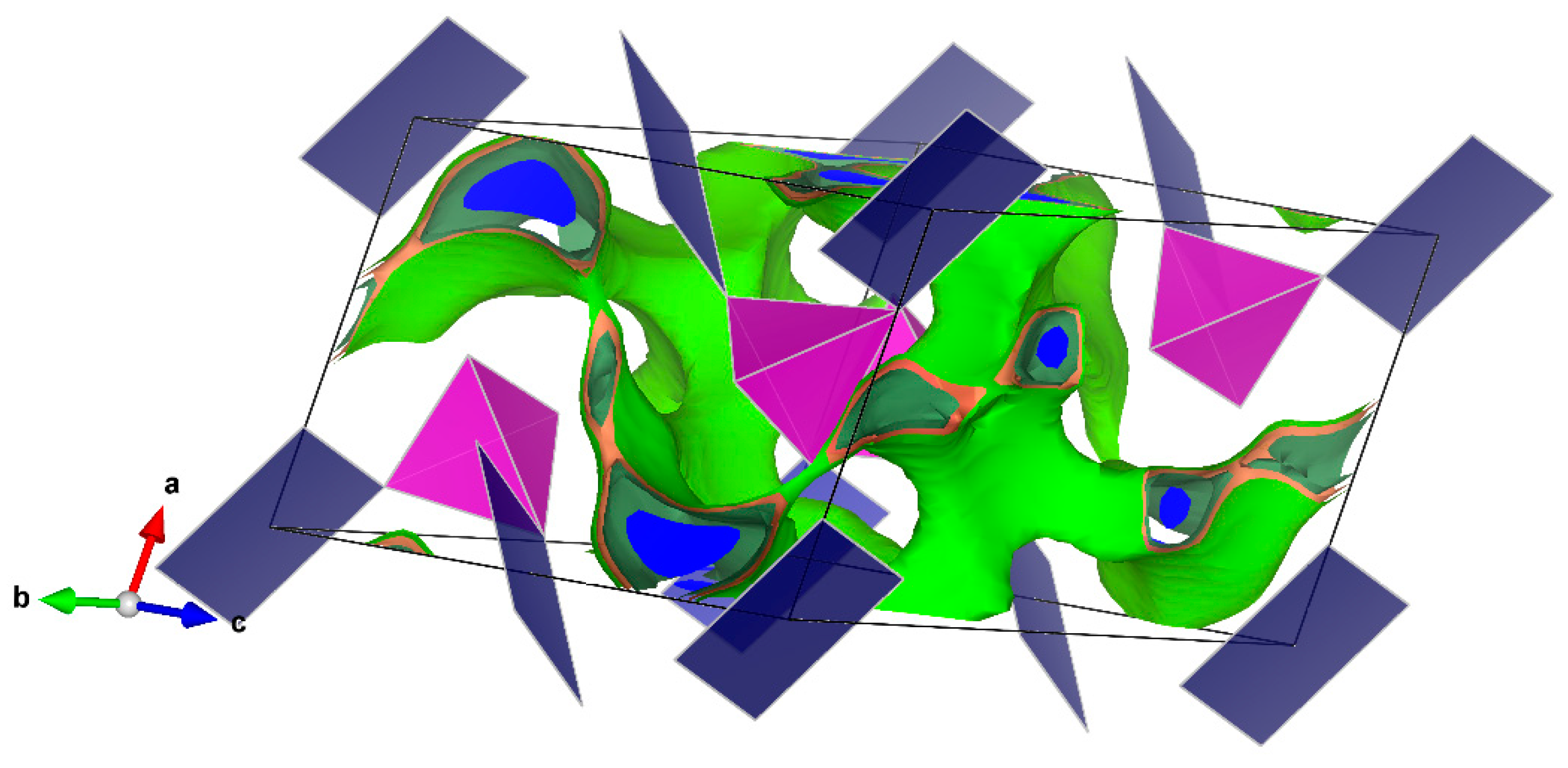
The bond-valence site energy (BVSE) model [
19,
20] was used to simulate the alkali migration in the 3D anionic framework. The BVSE model is the latest extension of the bond-valence sum (BVS) model developed by Pauling [
21] to describe the formation of inorganic materials. The BVS model was improved by Brown & Altermatt [
22] followed by Adams [
23], resulting in the expression:
where s
A-X is individual bond-valence, R
A-X is the distance between counter-ions A and X,
R0 and
b are fitted empirical constants, and
R0 is the length of a bond of unit valence.
The BVSE model was extensively used in the cation motion simulation in the anionic framework by following the valence unit as a function of migration distance [
24]. The valence unit was also recently related to potential energy scale and electrostatic interactions [
19,
20]. The BVSE method was used with success to simulate the transport pathways of monovalent cations (Na
+; K
+ and Ag
+) in numerous materials including Na
2CoP
1.5As
0.5O
7 [
15], Na
1.14K
0.86CoP
2O
7 [
25] and Ag
3.68Co
2(P
2O
7)
2 [
26]. The BVSE calculations were performed using the SoftBV [
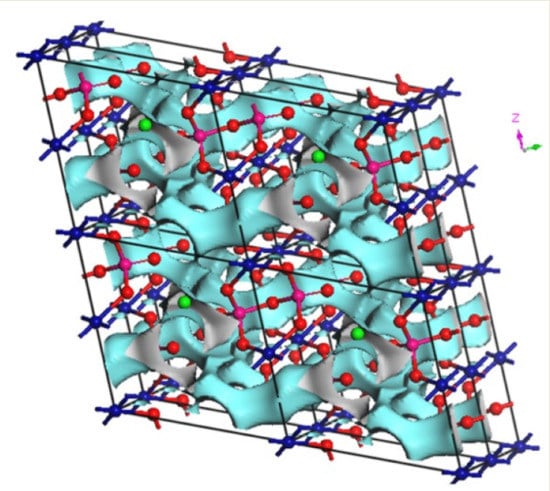
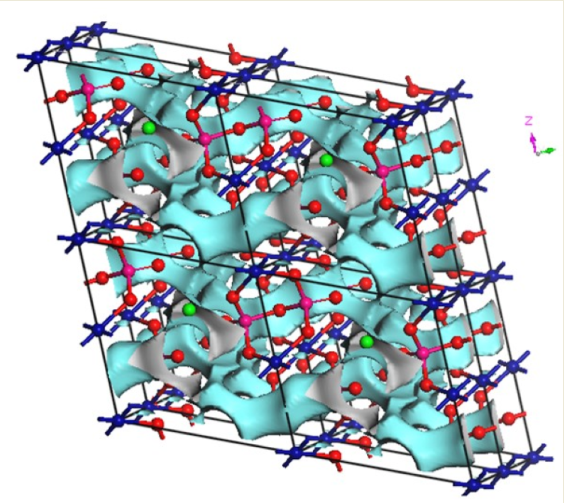
27] code and the visualization of isosurfaces was carried out using VESTA3 software (version 3, Koichi Momma and Fujio Izumi, 2018).

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}