Population-Based Parameter Identification for Dynamical Models of Biological Networks with an Application to Saccharomyces cerevisiae




Abstract
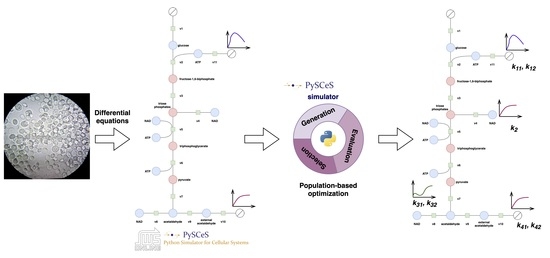
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- We provide a population-based optimization framework for parameter identification and showcase its performance on the example of the glycolysis of Saccharomyces cerevisiae, one of the most studied species in biology.
- We analyze the performance of the population-based optimization framework in the considered problem and indicate its high potential for future research.
- We extend the Python framework PySCeS [7] by implementing the population-based optimization methods (four methods known in the literature, and two new methods) in Python. The code for the methods together with the experiments is available online: https://github.com/jmtomczak/popi4sb.
2. Materials and Methods
2.1. Derivative-Free Optimization
2.2. Population-Based Optimization Methods
- (Init) Initialize and evaluate all individuals .
- (Generation) Generate new candidate solutions using the current population, .
- (Evaluation) Evaluate all candidates solutions:
- (Selection) Select a new population using the candidate solutions and the old populationGo to Generate or terminate.
2.2.1. Evolutionary Strategies (ES)
2.2.2. Differential Evolution (DE)
2.2.3. Reversible Differential Evolution (RevDE)
2.2.4. Estimation of Distribution Algorithms (EDA)
2.2.5. Population-Based Methods with Surrogate Models (RevDE+ & EDA+)
2.3. The Model of Glycolysis in Saccharomyces Cerevisiae
- Initial conditions
| . |
- Real parameter values
3. Experimental Setup
3.1. Implementation
3.2. Parameter Identification & the Fitness Function
3.3. Simulated Data
3.4. Settings
4. Results & Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ingalls, B.P. Mathematical Modeling in Systems Biology: An Introduction; MIT Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Nielsen, J. Systems biology of metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Ideker, T.; Galitski, T.; Hood, L. A new approach to decoding life: Systems biology. Annu. Rev. Genom. Hum. Genet. 2001, 2, 343–372. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Palsson, B.O. The evolution of molecular biology into systems biology. Nat. Biotechnol. 2004, 22, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Audet, C.; Hare, W. Derivative-Free and Blackbox Optimization; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Larson, J.; Menickelly, M.; Wild, S.M. Derivative-free optimization methods. arXiv 2019, arXiv:1904.11585. [Google Scholar] [CrossRef] [Green Version]
- Olivier, B.G.; Rohwer, J.M.; Hofmeyr, J.H.S. Modelling cellular systems with PySCeS. Bioinformatics 2005, 21, 560–561. [Google Scholar] [CrossRef] [Green Version]
- Olivier, B.G.; Snoep, J.L. Web-based kinetic modelling using JWS Online. Bioinformatics 2004, 20, 2143–2144. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.; Xu, R.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Duarte, N.C.; Herrgård, M.J.; Palsson, B.∅. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004, 14, 1298–1309. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Mensonides, F.I.; Brul, S.; Hellingwerf, K.J.; Bakker, B.M.; Teixeira de Mattos, M.J. A kinetic model of catabolic adaptation and protein reprofiling in Saccharomyces cerevisiae during temperature shifts. Febs. J. 2014, 281, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Yeast systems biology: Model organism and cell factory. Biotechnol. J. 2019, 14, 1800421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orij, R.; Urbanus, M.L.; Vizeacoumar, F.J.; Giaever, G.; Boone, C.; Nislow, C.; Brul, S.; Smits, G.J. Genome-wide analysis of intracellular pH reveals quantitative control of cell division rate by pH c in Saccharomyces cerevisiae. Genome Biol. 2012, 13, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.; Passarge, J.; Somsen, O.J.; Snoep, J.L.; Heinrich, R.; Westerhoff, H.V. Transduction of intracellular and intercellular dynamics in yeast glycolytic oscillations. Biophys. J. 2000, 78, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R.; Schonlau, M.; Welch, W.J. Efficient global optimization of expensive black-box functions. J. Glob. Optim. 1998, 13, 455–492. [Google Scholar] [CrossRef]
- Eiben, A.E.; Smith, J.E. Introduction to Evolutionary Computing; Springer: Berlin/Heidelberg, Germany, 2015; Volume 53. [Google Scholar]
- Gallagher, M.; Frean, M. Population-based continuous optimization, probabilistic modelling and mean shift. Evol. Comput. 2005, 13, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R.P.; De Freitas, N. Taking the human out of the loop: A review of Bayesian optimization. Proc. IEEE 2015, 104, 148–175. [Google Scholar] [CrossRef] [Green Version]
- Bäck, T.; Foussette, C.; Krause, P. Contemporary Evolution Strategies; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Moré, J.J.; Wild, S.M. Benchmarking derivative-free optimization algorithms. SIAM J. Optim. 2009, 20, 172–191. [Google Scholar] [CrossRef] [Green Version]
- Schwefel, H.P. Numerische Optimierung von Computer-Modellen Mittels der Evolutionsstrategie; Springer: Berlin/Heidelberg, Germany, 1977. [Google Scholar]
- Storn, R.; Price, K. Differential evolution—A simple and efficient heuristic for global optimization over continuous spaces. J. Glob. Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- Price, K.; Storn, R.M.; Lampinen, J.A. Differential Evolution: A Practical Approach to Global Optimization; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Pedersen, M.E.H. Good Parameters for Differential Evolution; Technical Report HL1002; Hvass Laboratories, 2010; Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.298.2174&rep=rep1&type=pdf (accessed on 4 January 2021).
- Tomczak, J.M.; Weglarz-Tomczak, E.; Eiben, A.E. Differential Evolution with Reversible Linear Transformations. arXiv 2020, arXiv:2002.02869. [Google Scholar]
- Larrañaga, P.; Lozano, J.A. Estimation of Distribution Algorithms: A New Tool for Evolutionary Computation; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Mühlenbein, H.; Paass, G. From recombination of genes to the estimation of distributions I. Binary parameters. In International Conference on Parallel Problem Solving from Nature; Springer: Berlin/Heidelberg, Germany, 1996; pp. 178–187. [Google Scholar]
- Pelikan, M.; Hauschild, M.W.; Lobo, F.G. Estimation of distribution algorithms. In Springer Handbook of Computational Intelligence; Springer: Berlin/Heidelberg, Germany, 2015; pp. 899–928. [Google Scholar]
- Jin, Y. Surrogate-assisted evolutionary computation: Recent advances and future challenges. Swarm Evol. Comput. 2011, 1, 61–70. [Google Scholar] [CrossRef]
- Hynne, F.; Danø, S.; Sørensen, P.G. Full-scale model of glycolysis in Saccharomyces cerevisiae. Biophys. Chem. 2001, 94, 121–163. [Google Scholar] [CrossRef]
- Kourdis, P.D.; Goussis, D.A. Glycolysis in saccharomyces cerevisiae: Algorithmic exploration of robustness and origin of oscillations. Math. Biosci. 2013, 243, 190–214. [Google Scholar] [CrossRef]
- Teusink, B.; Passarge, J.; Reijenga, C.A.; Esgalhado, E.; Van der Weijden, C.C.; Schepper, M.; Walsh, M.C.; Bakker, B.M.; Van Dam, K.; Westerhoff, H.V.; et al. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 2000, 267, 5313–5329. [Google Scholar] [CrossRef]
- Van Eunen, K.; Bouwman, J.; Daran-Lapujade, P.; Postmus, J.; Canelas, A.B.; Mensonides, F.I.; Orij, R.; Tuzun, I.; Van Den Brink, J.; Smits, G.J.; et al. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. FEBS J. 2010, 277, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://jjj.bio.vu.nl/models/wolf/ (accessed on 7 August 2020).
- Cranmer, K.; Brehmer, J.; Louppe, G. The frontier of simulation-based inference. Proc. Natl. Acad. Sci. USA 2020, 117, 30055–30062. [Google Scholar] [CrossRef] [PubMed]
- Gatopoulos, I.; Lepert, R.; Wiggers, A.; Mariani, G.; Tomczak, J. Evolutionary Algorithm with Non-parametric Surrogate Model for Tensor Program optimization. In Proceedings of the 2020 IEEE Congress on Evolutionary Computation (CEC), Glasgow, UK, 19–24 July 2020. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weglarz-Tomczak, E.; Tomczak, J.M.; Eiben, A.E.; Brul, S. Population-Based Parameter Identification for Dynamical Models of Biological Networks with an Application to Saccharomyces cerevisiae. Processes 2021, 9, 98. https://doi.org/10.3390/pr9010098
Weglarz-Tomczak E, Tomczak JM, Eiben AE, Brul S. Population-Based Parameter Identification for Dynamical Models of Biological Networks with an Application to Saccharomyces cerevisiae. Processes. 2021; 9(1):98. https://doi.org/10.3390/pr9010098
Chicago/Turabian StyleWeglarz-Tomczak, Ewelina, Jakub M. Tomczak, Agoston E. Eiben, and Stanley Brul. 2021. "Population-Based Parameter Identification for Dynamical Models of Biological Networks with an Application to Saccharomyces cerevisiae" Processes 9, no. 1: 98. https://doi.org/10.3390/pr9010098
APA StyleWeglarz-Tomczak, E., Tomczak, J. M., Eiben, A. E., & Brul, S. (2021). Population-Based Parameter Identification for Dynamical Models of Biological Networks with an Application to Saccharomyces cerevisiae. Processes, 9(1), 98. https://doi.org/10.3390/pr9010098