
MRlogP: Transfer Learning Enables Accurate logP Prediction Using Small Experimental Training Datasets



Abstract
:1. Introduction
1.1. Prediction of logP
1.2. Motivation for This Study
2. Materials and Methods
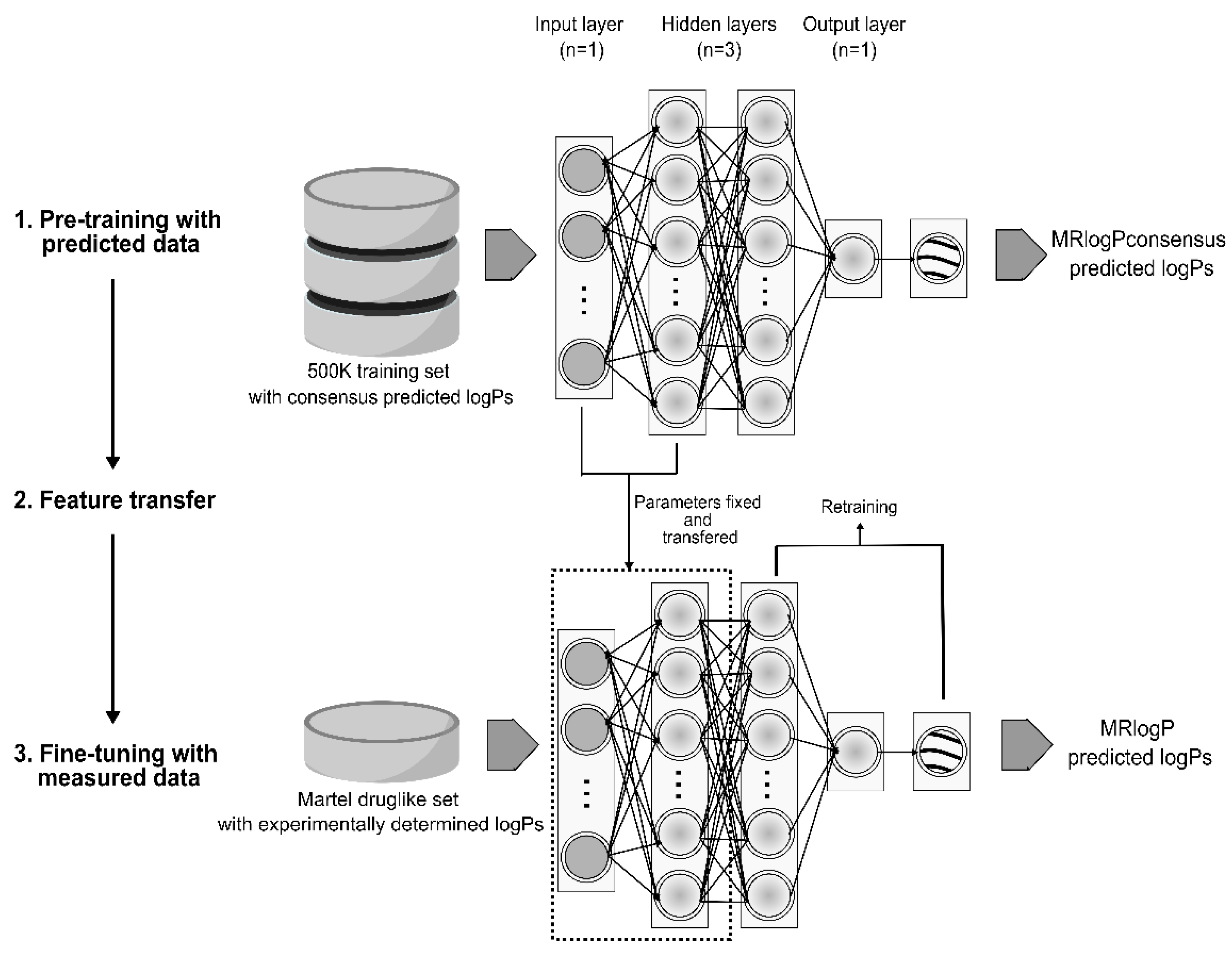
2.1. Dataset Preparation
2.2. Neural Network Architecture and Hyperparameters
3. Results
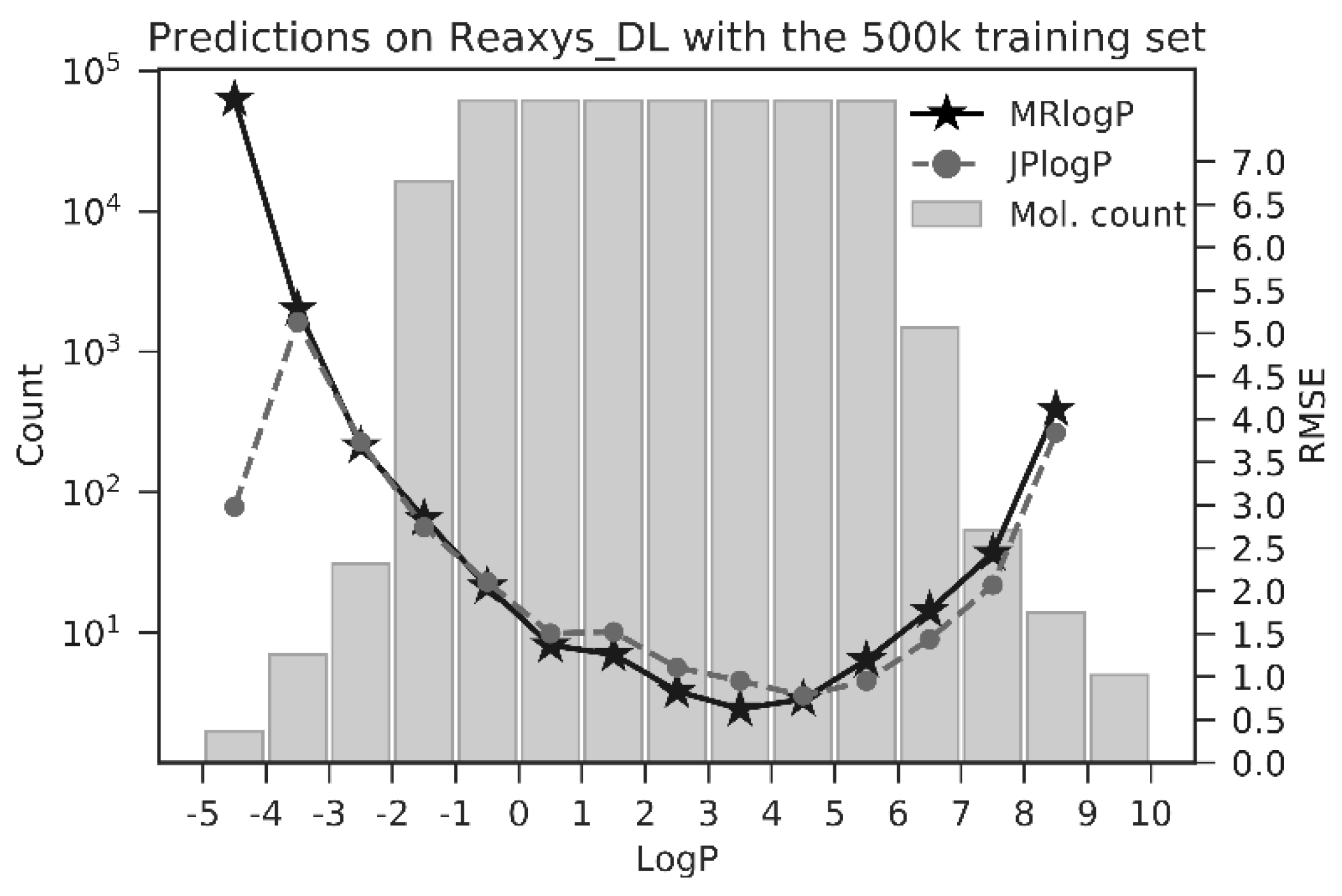
3.1. Arteficial Neural Network Training and Validation on Consensus logPs
3.2. Transfer Learning
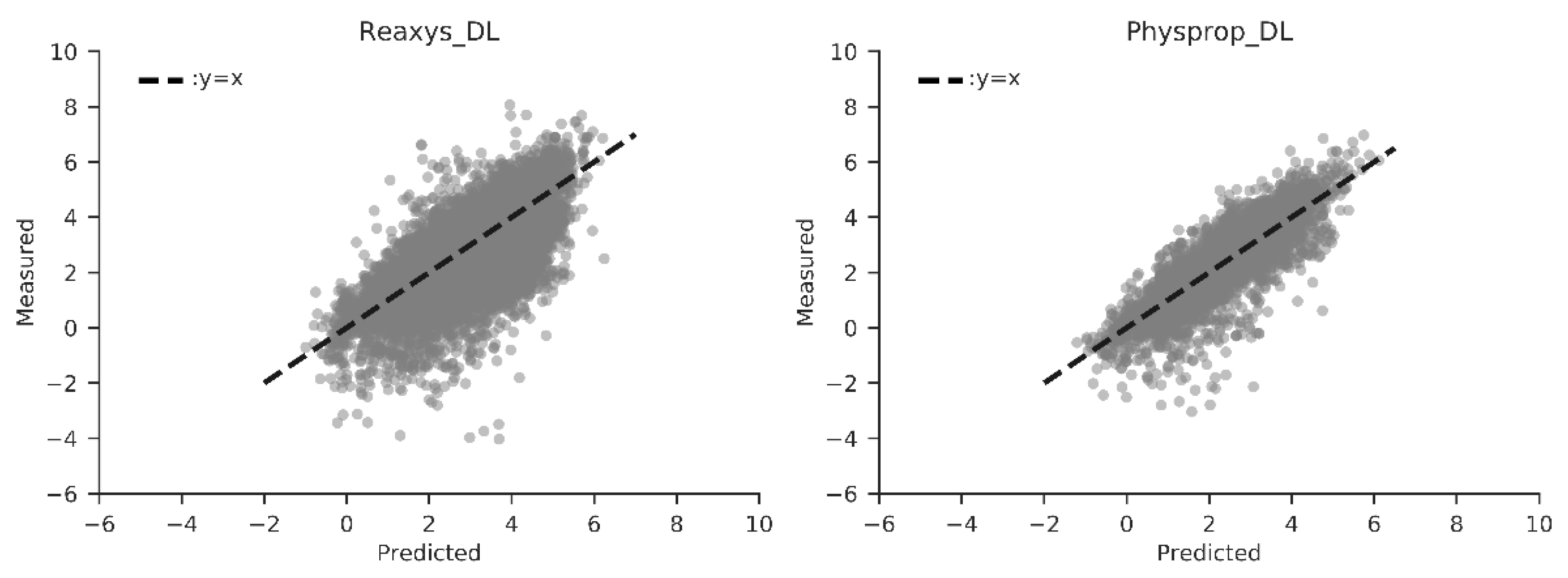
3.3. Performance Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharm. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Oprea, T.I. Current trends in lead discovery: Are we looking for the appropriate properties? J. Comput. Aided. Mol. Des. 2002, 16, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Allu, T.K.; Fara, D.C.; Rad, R.F.; Ostopovici, L.; Bologa, C.G. Lead-like, drug-like or “Pub-like”: How different are they? J. Comput. Aided. Mol. Des. 2007, 21, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Sangster, J. Octanol-Water Partition-Coefficients of Simple Organic-Compounds. J. Phys. Chem. Ref. Data 1989, 18, 1111–1229. [Google Scholar] [CrossRef]
- Moerlein, S.M.; Laufer, P.; Stocklin, G. Effect of lipophilicity on the in vivo localization of radiolabelled spiperone analogues. Int. J. Nucl. Med. Biol. 1985, 12, 353–356. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert. Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef]
- Mannhold, R.; van de Waterbeemd, H. Substructure and whole molecule approaches for calculating logP. J. Comput. Aided. Mol. Des. 2001, 15, 337–354. [Google Scholar] [CrossRef]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol− water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Plante, J.; Werner, S. JPlogP: An improved logP predictor trained using predicted data. J. Cheminformatics 2018, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, I.; HIRONO, S.; LIU, Q.; NAKAGOME, I.; MATSUSHITA, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. Model. 2002, 21, 47–49. [Google Scholar] [CrossRef]
- Goss, K.-U. Predicting the equilibrium partitioning of organic compounds using just one linear solvation energy relationship (LSER). Fluid Phase Equilibria 2005, 233, 19–22. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of logP methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]
- Wang, R.; Fu, Y.; Lai, L. A new atom-additive method for calculating partition coefficients. J. Chem. Inf. Comput. Sci. 1997, 37, 615–621. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Mansouri, K.; Grulke, C.M.; Judson, R.S.; Williams, A.J. OPERA models for predicting physicochemical properties and environmental fate endpoints. J. Cheminformatics 2018, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Martel, S.; Gillerat, F.; Carosati, E.; Maiarelli, D.; Tetko, I.V.; Mannhold, R.; Carrupt, P.A. Large, chemically diverse dataset of logP measurements for benchmarking studies. Eur. J. Pharm. Sci. 2013, 48, 21–29. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Soliman, K.; Grimm, F.; Wurm, C.A.; Egner, A. Predicting the membrane permeability of organic fluorescent probes by the deep neural network based lipophilicity descriptor DeepFl-LogP. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 23 July 2021).
- Saubern, S.; Guha, R.; Baell, J.B. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminformatics 2012, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely Available Conformer Generation Methods: How Good Are They? J. Chem. Inf. Model. 2012, 52, 1146–1158. [Google Scholar] [CrossRef]
- Lawson, A.J.; Swienty-Busch, J.; Géoui, T.; Evans, D. The making of reaxys—towards unobstructed access to relevant chemistry information, in The Future of the History of Chemical Information. In The Future of the History of Chemical Information; ACS Publications: Washington, DC, USA, 2014; pp. 127–148. [Google Scholar]
- Xu, B.; Wang, N.; Chen, T.; Li, M. Empirical evaluation of rectified activations in convolutional network. arXiv 2015, arXiv:1505.00853. [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo-and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Predictor | Performance (RMSE) | ||
|---|---|---|---|
| Martel_DL (N = 244) | Reaxys_DL (N = 20,067) | Physprop_DL (N = 5638) | |
| MRlogPconsensus | 0.972 | 1.074 | 0.727 |
| MRlogP | - | 0.988 | 0.715 |
| JPlogP | 1.007 | 1.196 | 0.738 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-K.; Shave, S.; Auer, M. MRlogP: Transfer Learning Enables Accurate logP Prediction Using Small Experimental Training Datasets. Processes 2021, 9, 2029. https://doi.org/10.3390/pr9112029
Chen Y-K, Shave S, Auer M. MRlogP: Transfer Learning Enables Accurate logP Prediction Using Small Experimental Training Datasets. Processes. 2021; 9(11):2029. https://doi.org/10.3390/pr9112029
Chicago/Turabian StyleChen, Yan-Kai, Steven Shave, and Manfred Auer. 2021. "MRlogP: Transfer Learning Enables Accurate logP Prediction Using Small Experimental Training Datasets" Processes 9, no. 11: 2029. https://doi.org/10.3390/pr9112029
APA StyleChen, Y. -K., Shave, S., & Auer, M. (2021). MRlogP: Transfer Learning Enables Accurate logP Prediction Using Small Experimental Training Datasets. Processes, 9(11), 2029. https://doi.org/10.3390/pr9112029